





Abstract
Cystic fibrosis (CF) is a complex disease which requires interdisciplinary care in specialized CF centres. In Germany, 97 paediatric and adult outpatient clinics agreed to report clinical data of their patients to a newly established registry, the Cystic Fibrosis Quality Assurance (CFQA) project. This article characterizes the design of the CFQA and the health status of the patients enrolled by the end of 1997.
Data from 4,306 patients reported to the CFQA project were analysed. Nutritional status and lung function of the patients were examined as well as the use of specific therapeutic interventions.
Mean age of all 4,182 patients alive by the end of 1997 was 15.7 yrs (maximum, 58 yrs), and 35.8% of patients were >18.0 yrs of age. One-third of the CF population were treated in the nine largest centres (each caring for >100 patients). Abnormal nutritional status (weight-for-height >90% of predicted or body mass index <19.0 kg·m2, respectively) was observed in 26.8% of children and adolescents and in 38.3% of adults. Lung function was abnormal (forced expiratory volume in one second <80% predicted) in the majority of adults (83.9%) and in 42.5% of the younger patients. The mortality rate was 1.4 of 100 patients in 1997. No clear association of clinical status with centre size was observed.
The clinical features of patients treated in German cystic fibrosis centres were generally comparable to those reported from other countries, although improvements are certainly warranted. The Cystic Fibrosis Quality Assurance project represents an important tool for future progress in the quality of cystic fibrosis care.
This work was supported by Christiane Herzog Stiftung, Mukoviszidose e.V., and Niedersächsischer Verein zur Förderung der Qualität im Gesundheitswesen.
Cystic fibrosis (CF) is the most common lethal inherited disease of the Caucasian population affecting ∼1 in 2,500 live births. The general dysfunction of exocrine glands leads to impaired function of many different organ systems, of which the respiratory tract and the exocrine pancreas are of prime clinical relevance. Due to improved conservative therapy, most patients now survive into adulthood, and CF is no longer exclusively a childhood disease. In recent years it became evident that specialized care in CF outpatient clinics and centres is of major importance in order to achieve optimum health of all patients 1–3. A multiprofessional approach with dieticians, respiratory therapists, psychologists, specialized CF nurses and physicians is provided in many CF centres. In some countries, standards for CF centres regarding personnel and equipment have been established, and CF centres have been accredited by national CF societies.
In Germany, the quality of CF care has become an important issue. Before the unification of East and West Germany in 1990, patients were treated in >100 outpatient clinics and private practices which had not been accredited. Basic patient data were reported once a year to two different registries in East or West Germany. In 1995, a decision was made to improve the quality of CF care by establishing the Cystic Fibrosis Quality Assurance (CFQA) project. The aims were to improve the clinical management and CF care in Germany with regard to quality of structure (centres), process (evaluation and treatment), and results (medical data) 4. The objective was to provide a framework to characterize longitudinally the clinical course of CF patients in relation to different conditions and relevant risk factors 5. The project aimed to generate comparative data allowing caregivers to evaluate their results in relation to those of other centres as a benchmark in order to learn from the best. In addition, criteria for CF clinics were defined, and the process of their accreditation has now been started.
The results are presented from a cross-sectional analysis of morbidity and mortality of CF patients in Germany as recorded in the central CFQA database in 1997. These data could serve as a basis for future reports and as a framework for analyses of special topics relevant for CF.
Materials and methods
Cystic Fibrosis Quality Assurance Project
The CFQA project was founded in 1995 and was located at the Centre for Quality Management in the Health Care System in Hannover. The German CF Foundation (Mukoviszidose e. V.) agreed to sponsor the initial phase of the project.
Clinical record forms were developed specifically for this project, incorporating experiences from previous CF databases. The basic sheet contains demographic data, genotype, date of CF diagnosis and initial symptoms. Yearly follow-up sheets collect information on social situation (marital status, housing situation, further siblings, schooling and professional training), current medical problems associated with CF (e.g. pneumothorax, diabetes mellitus, hepatobiliary complications), clinical data (weight, height, Shwachman score), pulmonary function (vital capacity (VC), forced expiratory volume in one second (FEV1) and midexpiratory flow at 25% of vital capacity (MEF25)), serum immunoglobulin-(Ig)G and microbiology, as well as data on treatment regimens such as use of antibiotics, oral antidiabetics or dose of pancreatic enzymes. Medical complications like allergic bronchopulmonary aspergillosis (ABPA), massive haemoptysis, hepatobiliary disease and CF related diabetes as well as distal intestinal obstruction syndrome (DIOS) were defined according to current criteria 5.
In order to guarantee the protection of patients' personal data, each patient was assigned a unique identifier. This was patient-specific in order to preclude duplicate entry of data. Patient names were not transferred to the CFQA project; each CF centre holds lists with the respective coded patient identifiers. Written informed consent was obtained by parents and/or patients.
The initial phase of the project started in 1995. Each of the 111 centres known to the German CF Foundation was invited to participate. Centres reported patient data once a year from a routine visit near the patient's birthday when the patient was in a stable clinical condition. Patient record forms were mailed to the CFQA database manager in Hannover once a year. A cross-sectional analysis of data obtained in 1997 are presented in this article.
Database and statistical analysis
A database was programmed in Microsoft Access 97 (Microsoft GmbH, Unterschleißheim, Germany). Plausibility tests were programmed for the identification of incorrect or inconsistent data (e.g. FEV1 must not be greater than the forced vital capacity (FVC)). For statistical analysis, data were transferred into the Statistical Package for Social Sciences (SPSS) software version 7.5 (SPSS Inc., Chicago, IL, USA). The percentage weight-for-height was calculated using normal values from German children 6. Lung function reference values were selected according to the recommendations of the European Coal and Steel Community 7, 8. Z-scores for height, weight and serum IgG were calculated using appropriate age-related reference values 9.
For the analysis of outcome variables, values were defined as being in the desired range of quality, i.e. considered normal according to the following limits: weight-for-height ≥90% of predicted in children and adolescents, body mass index (BMI)≥19 kg·m2 in adults, FVC≥80%, FEV1≥80% and MEF25≥60% of pred. Serum IgG values were considered increased if Z-scores were >+2. Height and weight were abnormal if Z-scores were <−2.
Unpaired t-tests were used for the comparison of patient groups, e.g. for means of lung function values in children versus adults. Means of ≥3 different groups (e.g. age groups or types of CF centres) were compared using parametric or nonparametric analysis of variance. The Chi-squared test was used to determine differences between groups regarding frequencies of certain variables. p-Values<0.05 were considered statistically significant.
Results
Cystic fibrosis centres
Of the 111 centres invited to participate, 97 centres caring for 4,306 patients had agreed to take part by December 31, 1997. The majority of the remaining 14 centres were small adult CF clinics with less than five patients, and only three centres were assumed to care for >20 CF patients. Patient numbers differed considerably between centres (table 1⇓). The nine largest institutions (each caring for >100 patients) covered 36% of all patients registered. The majority of patients were being treated at one of the medium sized centres (21–100 patients). Outpatient clinics at university hospitals were attended by 60% of patients. For adult care, 15 centres had been established by end of 1997, and these were attended by 20.8% of all CF subjects>18.0 yrs.
Cystic Fibrosis centres and patient demography
Patient demography
Data from 4,306 patients were reported to the CFQA project between 1995 and December 31, 1997, of whom 4,182 were alive at that date (table 1⇑). Permission to participate could not be obtained from 133 patients (3.1%). Age distribution of the patients is shown in figure 1⇓. Mean age was 15.7 yrs (sd±9.4, median 14.4 yrs) with a range of 0–58 yrs, and 35.7% of patients were ≥18 yrs. There was a slight preponderance of males (53.1%). Sex distribution was not different between large, medium and small centres.

Sex distribution by age of the 4,181 patients alive at December 31, 1997 (sex of one patient missing). ▒: males, n=2,221; ▓: females, n=1,960.
Cystic fibrosis mutations
From the group of 3,070 patients who were genotyped (71.3%), both mutations were identified in 67.8% and one mutation in 24.6% of patients, respectively. The CF mutation remained unidentified in 20% of chromosomes and in 7.6% of patients. As shown in table 2⇓, ΔF508 deletion was the most frequent mutation (68.4%). Patients who were homozygous for this defect comprised the largest genotype group (50.1%), 36.5% of patients were compound heterozygotes for the ΔF508 mutation (ΔF508 plus another known CF mutation: 15.4%, ΔF508 plus an unknown CF mutation: 21.1%).
Frequencies of cystic fibrosis transmembrane regutar mutations in the chromosomes of 3,070 patients genotyped
New cystic fibrosis diagnoses
In 1997, 141 patients were newly diagnosed with CF. Median age at diagnosis was 7 months. Only 57% of patientspt patients were identified during the first year of life, and 6.4% were adults, the maximum age at diagnosis being 43 yrs. Meconium ileus was present in 18.4% of patients. Other clinical symptoms were: a combination of gastrointestinal and respiratory symptoms (27%), only gastrointestinal (26%) or only respiratory symptoms (21%), rectal prolapse (3.5%) or being the sibling of a CF patient (3.5%). Although neonatal screening for CF has been abandoned as a routine procedure in Germany, 11% of patients were diagnosed after a positive screening test.
Outcome variables
The data on outcome variables are from 3,448 different patients (table 3⇓), since follow-up records from 787 patients (21%) who had been reported in 1995 or 1996 were not available in 1997.
Clinical data in children and adults
Nutritional status
Means of weight-for-height in children and BMI in adults were within the normal range with values of 97.2% pred and 19.8 kg·m2, respectively. However, the proportion of patients who were underweight increased from 22% at <6 yrs to 25% from 6–12 yrs and further to 31% in adolescents (figs. 2 and 3⇓⇓). In the adult age group, 38% of patients were malnourished as indicated by a BMI<19 kg·m2. Adolescent and adult females had significantly higher weights than males. Height was less abnormal than weight, as indicated by near normal Z-scores at all ages. In general, height Z-scores increased with age, and normal means were present in the adult age group.

Boxplots of weight-for-height (expressed as per cent of predicted normal values) in children and adolescents with cystic fibrosis. The box represents the 25th–75th percentile range, the solid horizontal line the median (50th percentile), and the error bars the 10th and 90th percentiles, respectively. The number of patients for each age group in turn: <2: 145; <4: 215; <6: 242; <8: 283; <10: 289; <12: 294; <14: 296; <16: 279; <18: 282.

Boxplots of body mass index (kg·m2) in adult cystic fibrosis patients. The box represents the 25th–75th percentile range, sollid horizontal line the median (50th percentile), and the error bars the 10th and 90th percentiles, respectively. The number of patients for each age group in turn: <20: 201; <22: 158; <24: 129; <28: 247; <32: 212; <36: 95; >36: 68.
Pulmonary function
Results of pulmonary function tests from children >6.0 yrs of age who were able to perform reliable tests are presented in figures 4 and 5⇓⇓. Children and adolescents had mean values for FVC and FEV1 at the lower limit of normal, whereas mean MEF25 was abnormal (62.7% of pred). A significant proportion of children aged 6–8 yrs already had impaired respiratory function: FVC was abnormal in 30%, FEV1 in 21% and MEF25 in 28%. The proportion of patients with pathological values increased with advancing age. Adults had significantly worse respiratory function compared to children (p<0.001), with decreased means of all three parameters. Means of FVC and FEV1 were significantly better in larger centres in children (p<0.01), whereas the opposite was true for MEF25; this parameter of small airways obstruction was significantly worse in patients treated in large centres (p<0.01).
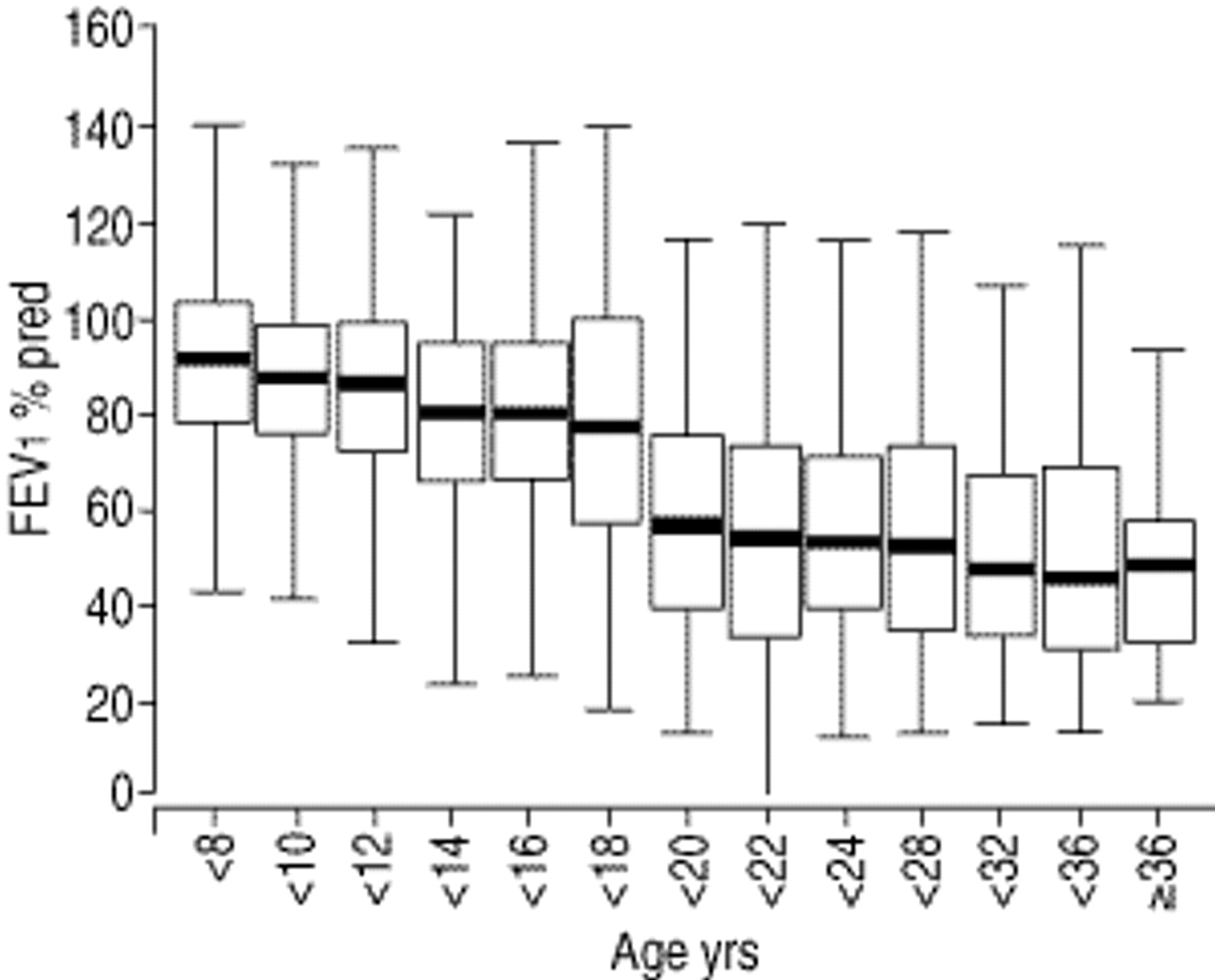
Boxplots of forced expiratory volume in one second (FEV1 % predicted) by age. The decline at age 18 is due to different reference values for adolescents and adults. The box represents the 25th–75th percentile range, the solid horizontal line the median (50th percentile) and the error bars the 10th and 90th percentiles, respectively. The number of patients for each age group in turn: <8: 234; <10: 273; <12: 285; <14: 286; <16: 263; <18: 271; <20: 196; <22: 154; <24: 125; <28: 238; <32: 207; <36: 88; ≥36: 62.

Boxplots of midexpiratory flow at 25% of vital capacity (MEF25 % predicted) by age. The decline at age 18 is due to different reference values for adolescents and adults. The box represents the 25th–75th percentile range, the solid horizontal line the median (50th percentile) and the error bars the 10th and 90th percentiles, respectively. The number of patients for each age group in turn: <8: 236; <10: 273; <12: 277; <14: 283; <16: 256; <18: 263; <20: 189; <22: 140; <24: 114; <28: 218; <32: 187; <36: 80; ≥36: 55.
Microbiology and serum immunoglobulin-G
Colonization with Pseudomonas aeruginosa was frequent in all age groups (43.7% in children and adolescents and 76.4% in adults). Younger children were significantly less frequently colonized with this bacterium (fig. 6⇓). Higher colonization rates were obtained in larger CF centres (p<0.01). Only few patients (2.1%) were colonized with Burkholderia cepacia. Abnormally elevated serum IgG values were found in 31.6% of patients <18 yrs of age and in 57.4% of adult patients, indicating a larger stimulation of the immune system with advancing age due to chronic bacterial infection.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histogram of the percentage of patients with positive cultures for Pseudomonas aeruginosa (└) or Burkholderia cepacia (□) at all ages.
Medical complication rates
The proportion of patients experiencing complications of their disease is summarized in table 4⇓. As expected, a significantly larger number of complications was reported in the adult age group. This was particularly true for ABPA, massive haemoptysis and diabetes mellitus. The most frequent respiratory complication was ABPA followed by surgery for nasal polyps. Regarding gastrointestinal complications, hepatobiliary disease was most frequent and affected 15.4% of all patients. Diabetes mellitus was quite common in adults, whereas intestinal obstruction showed no age dependency. Other concomitant diseases occurred in 13.3% of patients; however, no differentiation as to underlying diagnoses (e.g. asthma, arthritis, inflammatory bowel disease) was possible within the CFQA project.
Percentages of medical complication rates in 3,448 patients
Treatment
An important feature of the CFQA database is that it contains data on specific treatment modalities, which are listed in table 5⇓. The participating centres were encouraged to comply with current recommendations for CF care although treatment was left to the discretion of the treating physician. Basically, routine treatment consisted of inhalations with physiological saline and/or salbutamol, chest physiotherapy (clapping and postural drainage in young patients and autogenic drainage, positive expiratory pressure mask or flutter device in older patients), regular physical activity, high-energy meals, pancreatic enzymes and vitamin supplementation. In addition to baseline therapy, the majority of patients were treated with oral antibiotics. Some German centres prescribe prophylactic antistaphylococcal treatment, whereas others treat Staphylococcal aureus infection only when present in sputum or throat swab. Nearly 30% of patients inhaled antipseudomonal antibiotics, and 1,372 patients (45.2%) received at least one course of intravenous antipseudomonal antibiotics in 1997. Regarding gastrointestinal manifestations of the disease, only 7% were not taking pancreatic enzymes, and 20.3% of patients received >10,000 lipase units per kg·day−1. Ursodesoxycholic acid was also frequently prescribed. As expected, insulin was required by a considerable proportion (13.1%) of adults, however infrequently used in children and adolescents. Less than 5% of all patients received oral antidiabetics or nocturnal gastrostomy feedings.
Percentages of patients receiving specific treatment regimens
As expected, a considerably larger proportion of adult patients compared to children, inhaled Dornase alfa or received anti-inflammatory treatment (including oral steroids and nonsteroidal anti-inflammatory drugs), oxygen supplementation or assisted ventilation.
Mortality rate
In 1997, 27 males and 20 females with CF died, corresponding to a crude mortality rate of 1.4 in 100 patients. Mean and median age at death was 21.9 yrs with a range of 3–34 yrs. Lung function was severely compromised (FEV1 34.5% and MEF25 15.0% of pred normal values), weight was considerably reduced (mean 87% of normal weight-for-height in children, mean BMI 18.1 kg·m2 in adults), and nearly all patients were colonized with P. aeruginosa (94%). The major cause of death was stated as “cardiorespiratory” (78%), whereas “other CF relevant reasons” were mentioned in 18%, and “hepato-intestinal” or “non-CF” causes in 7% each, respectively.
Lung transplantation was performed in 28 patients between 1995–1997. Regular follow-up data from these patients will be included in updated versions of the clinical record forms of the CFQA project.
Discussion
The establishment of the CFQA project represents a major step forward towards providing optimum care for all CF patients in Germany. For the first time it is possible to compare the health status of the patients and treatment modalities between centres and with registries of other countries. In contrast to other databases such as the European CF Registry, the results are representative for the situation in Germany since virtually all CF clinics participate in the submission of patient data. The annual CFQA report anonymously presents centre data, allowing the CF teams to evaluate their own results against those of others and to improve their internal quality management. Also, comparisons with data obtained in previous years are possible in order to assess any progress made. These are important steps towards quality management in clinical CF care.
In Phase I of the project, centres were required to perform routine diagnostic tests such as respiratory function at least once a year. Although most CF centres in Germany use a scheme of routine diagnostics in CF, some centres have either not performed or not documented all of the required investigations. This is reflected by the proportion of missing values: data on respiratory function were not available in up to 9% of patients (MEF25), and serum IgG values were missing in 14.5%. A comprehensive documentation of all relevant data obtained at visits to the outpatient clinic will be performed in Phase II of the CFQA project. This requires an even larger discipline by the CF caregivers and certainly needs organizational changes in the outpatient clinic, e.g. by using electronic patient records during outpatient visits. Phase II has started with a pilot group of nine centres covering ∼800 patients, and depending on the experiences made it will be extended to all German CF centres. The ultimate goal of Phase II will be to “learn from the best” and to generate hypotheses about therapies with superior outcome which can then be tested in prospective, controlled studies. Clinical guidelines shall be established by incorporating results from these analyses.
When the present results are compared with published data from other national CF patient registries, it must be kept in mind that different reference values are used across publications. In the US patient registry 10 reference equations by Knudson et al. 11 were used for the evaluation of expiratory flow/volume curves. These have the advantage of being valid for all ages between 6–>70 yrs. In contrast, the authors used separate reference values for the paediatric 8 and for the adult age group >18.0 yrs 7. This leads to a sudden fall in percentage FEV1 pred of ∼10% at 18 yrs. Nevertheless, mean values for FEV1 and VC were in a comparable range in Germany (71.4 and 79.4% of pred in 1997), the USA (72.3 and 84.5% of pred in 1996), and in France (68.9% and 80.9% of pred in 1995), respectively 12.
The median age of patients was higher in Germany (14.4 yrs) and in the USA (13.8 yrs) than in France (11.6 yrs), and the crude mortality rate in Germany of 1.4 in 100 patients was low compared with that of the USA (1.9%) and France (2%). When interpreting these figures it must be taken into consideration that not all deaths might have been reported to the CFQA project. Yearly follow-up data from 787 patients who had been reported in previous years were unavailable in 1997, and some of these missing patients might have died. Furthermore, the proportion of patients with CF in relation to the whole population of ∼80 million is relatively low in Germany compared with the UK (6,500 CF patients) or the USA (20,000 patients). Whether this means that many patients remain undiagnosed in Germany or whether physicians in private practices do not transfer patients to specialized CF centres cannot be answered at present. To assure a complete database is one of the major tasks of the quality assurance project, and further progress is needed in this respect in the forthcoming years.
Aspects of treatment can also be compared between countries. Dornase alfa was prescribed in 40% of USA and in 23.5% of German patients, and anti-inflammatory agents in 5.4% of USA and in 6.5% of German patients. The proportion of patients receiving gastrostomy feedings was higher in the USA (6% versus 1.8% in Germany), and the same was true for home oxygen therapy (7% versus 2.7% in Germany). This points towards a more conservative therapeutic approach in Germany. A large difference was observed in prescription rates of ursodesoxycholic acid; only 0.8% of USA in contrast to 31.4% of German patients received this drug, suggesting that persistently increased liver enzymes or abnormal liver echogenicity on abdominal ultrasound are treated with ursodesoxycholic acid in Germany, but not in the USA.
Complication rates were more or less comparable between Germany and the USA with respect to massive haemoptysis, diabetes mellitus or DIOS. In contrast, ABPA was documented in 6.1% of German patients compared to only 1.9% in the USA, and the incidence of nasal polyps requiring surgery was also higher in Germany with 4.6% compared to only 2.5% in USA patients. Whether this is due to real differences in complication rates or merely reflects different diagnostic and therapeutic approaches cannot be evaluated from the available data.
An important difference to the situation in the USA is the fact that the process of accreditation of centres by an expert panel from the German CF Foundation has only recently been started in Germany. The large proportion of small outpatient clinics (n=40 of a total of 97 centres) caring for <20 patients reflects that the term “CF centre” was not defined so far, and it was not required to have a team of professional CF caregivers and certain equipment available. In the USA, 20,886 CF patients from 116 centres are reported in 1996, i.e. ∼180 patients per centre, whereas the corresponding figures in Germany and France are only 44 and 37 patients per centre, respectively. In 1999, certification of German CF centres was introduced including centre size >20 patients, participation at quality assurance, full medical inpatient and outpatient service including physiotherapy, dietary counselling, psychosocial support. The small centres in Germany will ultimately disappear in the future. From the relatively crude data reported here, a tendency towards worse lung function results was observed in small centres. A recent publication 3 has shown that centre care improves outcome in CF. As long as an allocation bias cannot be ruled out in the present study, these figures must be interpreted with caution. Further analyses are currently being carried out to investigate the influence of centre size on outcome more thoroughly. Quality indicators and determinants of prognosis like stable nutrition and lung function data and absence of P. aerigunosa infection over a long time period are being used for further quality management. Long-term follow-up data will only be available in the next few years.
During the last 5 yrs, the CFQA project has contributed significantly to the quality of CF patient care in Germany. CF centres regularly report patient data into the registry, and can compare their results with those of other centres by means of the yearly report. Statisticians at the documentation centre have implemented measures of plausibility to assure the quality of data. A standardized statistical analysis has been developed allowing to produce yearly reports of similar structure and completeness. Criteria for certification of CF centres have been established. Of the criteria of quality in the study of Donabedian 4, “structure” and “outcome” have been adequately covered so far by the CFQA project, whereas the “process” aspect needs additional consideration. For the future of CF care in Germany, the project offers considerable improvement. Above all, the introduction of a special CF software programme, cystic fibrosis evaluation system (CFAS), which is based on the CFQA case report form, will allow staff from CF centres to continuously monitor the health status of their patients at each clinic or hospital visit. CFAS provides inbuilt-reports of lung function, nutritional status, treatment, and complications. Another element will be the generation of routine reports for the referring physician. Automatic data transfer from CFAS to the CFQA documentation centre for the yearly reports will facilitate completeness of data within the German registry. Special statistical multivariate analysis will be introduced to evaluate risk factors for mortality in more detail. Site-visits at clinics participating in Phase II will be performed by monitors from the documentation centre in the year 2000 to further improve data quality. Physicians plan to analyse the data with respect to “learn from the best”, and they will arrange consensus conferences for important treatment topics.
In summary, the Cystic Fibrosis Quality Assurance project is an important element of a comprehensive programme aimed at improving the quality of care for cystic fibrosis in Germany. Results have revealed that the clinical features of cystic fibrosis patients in Germany are comparable with those of other countries. Further reports will be generated in order to define subgroups of high-risk patients and to evaluate specific aspects of therapy in more detail in order to incorporate practical quality management into cystic fibrosis healthcare.
Acknowledgments
The authors gratefully acknowledge the large amount of work and effort put into this project by participants from 97 cystic fibrosis outpatient clinics. Their enthusiasm and cooperation is highly appreciated. The authors particularly acknowledge special support given by M. Corey (Toronto, Canada).
- Received June 19, 2000.
- Accepted January 3, 2001.
- © ERS Journals Ltd









