





Abstract
Asthma can be effectively treated by the use of bronchodilator therapies administered by inhalation. The objective of this study was to describe the dose-response relationship of combined doses of fenoterol hydrobromide (F) and ipratropium bromide (I) (F/I) delivered via Respimat®, a soft mist inhaler, and to establish the Respimat® dose which is as efficacious and as safe as the standard marketed dose of F/I (100/40 µg) which is delivered via a conventional metered dose inhaler (MDI).
In a double-blind (within device) cross-over study with a balanced incomplete block design, 62 patients with stable bronchial asthma (mean forced expiratory volume in one second (FEV1) 63% predicted) were randomized at five study centres to receive five out of eight possible treatments: placebo, F/I 12.5/5, 25/10, 50/20, 100/40 or 200/80 µg delivered via Respimat®; F/I 50/20 or 100/40 µg delivered via MDI.
Pulmonary function results were based on the per-protocol dataset, comprising 47 patients. All F/I doses produced greater increases in FEV1 than placebo. A log-linear dose-response was obtained for the average increase in FEV1 up to 6 h (AUC0–6 h) and peak FEV1 across the dose range administered by Respimat®. Statistically, therapeutic equivalence was not demonstrated between any F/I dose administered by Respimat® compared with the MDI. However 12.5/5 and 25/10 µg F/I administered via Respimat® were closest (slightly superior) to the F/I dose of 100/40 µg delivered via MDI. Pharmacokinetic data from 34 patients indicated a two-fold greater systemic availability of both drugs following inhalation by Respimat® compared to MDI. In general, the active treatments were well tolerated and safe with regard to vital signs, electrocardiography, laboratory parameters and adverse events.
In conclusion, combined administration of fenoterol hydrobromide and ipratropium bromide via Respimat®, is as effective and as safe as higher doses given via a metered dose inhaler.
National and international guidelines for the management of asthma recommend the inhalation of short-acting beta2-agonists, when required by the patient, as an initial bronchodilation therapy for acute severe asthma 1–6. In addition, this can be supplemented with anticholinergics where there is inadequate control of asthma 7.
The sympathomimetic agent, fenoterol hydrobromide (F), has a high potency and selectivity for beta2-adrenoreceptors and elicits bronchodilation by relaxing bronchial smooth muscle. Ipratropium bromide (I) has a different mechanism of action, acting as an anticholinergic agent at muscarinic receptors in the respiratory tract to relieve bronchoconstriction. The two compounds also differ in pharmacodynamics, with F having a rapid onset of action, causing prompt, short-acting bronchodilation. In contrast, I has a slower onset but prolonged duration of action. This complementary mechanism between F and I has led to their use in fixed dose combinations for several years [8–11].
There are currently a range of inhaler devices available for delivering antiasthma therapies 12. Metered dose inhalers (MDIs) are the most common, because of their safety, efficacy and ease of use. However, these devices use chlorofluorocarbon (CFC) propellants which are being withdrawn because of environmental concerns 13. There are alternatives for CFC-driven MDIs under development, some of which are already available. These include MDIs containing hydrofluoroalkane propellants and dry powder inhalers (DPIs) 14, 15, with each device having advantages and disadvantages based on lung deposition characteristics, reliability, consistency and ease of use. In addition, Respimat® (Boehringer Ingelheim KG, Ingelheim, Germany) has been developed: a reusable, mechanically-driven, propellant free, multi-dose, soft mist inhaler (SMI). Respimat® releases the drug solution as a soft mist over a period of 1.2 s, at a particle velocity (∼10 m·s−1) five times slower than from conventional MDIs 16. In addition, a high proportion of the drug dose is in the fine particle fraction (particles with diameter <5.8 µm). Scintigraphic studies have demonstrated that the smaller particle size and lower velocity of the dose from Respimat® improves lung deposition compared to conventional MDIs 17–23. Furthermore, clinically relevant bronchodilation has been observed in 2-way crossover pilot studies with a prototype SMI delivering either F or I, in asthmatics or chronic obstructive pulmonary disease patients 24, 25.
The present study was designed to assess the efficacy and safety of the Respimat® in delivering combined doses of F and I (F/I), compared with a conventional MDI, in patients with stable asthma.
Materials and methods
Patients
Patients eligible for the study were those aged 18–65 yrs, with stable perennial bronchial asthma as defined by the American Thoracic Society 26. At the initial screening visit, each patient provided written informed consent and underwent a complete medical examination to fulfil inclusion and exclusion criteria. Included patients had stable asthma with no hospital admission for an exacerbation and with no major change in medication for ≥6 weeks prior to the trial. All included patients had an initial forced expiratory volume in one second (FEV1) of 40–80% of the predicted normal value according to standard criteria 27, 28. In addition, all patients exhibited reversible airway obstruction as shown by an increase in FEV1 of ≥15% within 60 min after inhaling two puffs of 50/20 µg F/I via MDI without a spacer, following withdrawal of other bronchodilatory drugs. Furthermore, baseline FEV1 for each patient, on each test day, had to be within 20% and 0.3 L of that obtained on the first test day. Eligible subjects were nonsmokers or exsmokers who had given up smoking for ≥1 yr and with a history of no more than ten pack years. Subjects were excluded if they had a respiratory tract infection, severe exacerbation of asthma within 6 weeks prior to the trial, or were intolerant to the study drugs of ex-cipients. Patients were also ineligible if they were pregnant or lactating, receiving oral corticosteroids within 6 weeks prior to the study, or receiving beta-blockers. Appropriate withdrawal times were used for other pulmonary medications (antihistamines 48 h, inhaled short-acting beta2-sympathomimetics 8 h, inhaled long-acting beta2-sympathomimetics 48 h, inhaled anticholinergics 12 h, slow release xanthines 72 h). Use of inhaled cromolyn sodium/nedocromil and stable use of inhaled corticosteroids was permitted until 1 h before pre-dose evaluation of pulmonary function.
Sixty-two eligible patients were randomised. The data of eight patients from one test centre were excluded from efficacy and safety analyses because of major protocol violations. The remaining 54 patients were included in the safety analysis. Fifty patients were included in the intent-to-treat efficacy data set, since one patient withdrew after the first test day and three patients were excluded due to insufficient data. Following the early discontinuation of a further patient, and exclusion of two others with incomplete data, the per-protocol analysis was finally based on 47 patients without major protocol violations (table 1⇓).
Patient demographics and baseline characteristics
This trial was carried out in accordance with the principles of the declaration of Helsinki and approved by the Ethics Committee of the Federal Chamber of Physicians of Rhineland-Palatinate (Landesärztekammer von Rheinland-Pfalz).
Study design
In a randomised, five-period cross-over (balanced incomplete block) design at five study centres, included patients received five out of these eight possible treatments: placebo; F/I doses of 12.5/5, 25/10, 50/20, 100/40 or 200/80 µg delivered via Respimat®; or F/I doses of 50/20 or 100/40 µg delivered using a conventional MDI. On each test day, patients inhaled either one puff from Respimat® or two puffs delivered via MDI: either two puffs of 50/20 µg or one puff of 50/20 µg F/I plus one puff placebo MDI (all devices from Boehringer-Ingelheim KG). Treatment was open label between devices and double-blind within each device. Patients' pulmonary function (FEV1 and forced vital capacity (FVC)), adverse events and vital signs were evaluated until 6 h after inhalation as described below. Test days were ≥2 days apart and medication was inhaled at the same time on each test day ±0.5 h, between 07:00 h and 10:00 h.
Methods
Pulmonary function (FEV1, FVC) was measured by spirometry at baseline, then at 5, 15, 30, 60 and 90 min and at 2, 3, 4, 5 and 6 h after administration of the test drug on all test days. The primary endpoint was the average FEV1 in litres. This was determined as area under the curve for FEV1 change from test day baseline up to 6 h divided by 6 h (AUC0–6 h). Secondary endpoints used to assess patient bronchodilatory responses were the peak change in FEV1, time to onset and duration of the therapeutic response, and time to peak therapeutic response. A therapeutic response was defined as an FEV1 measurement exceeding the predose value by 15% at any time throughout the 6-hour observation period. Time to onset was defined as the linear interpolation of the time of the first therapeutic response and the time of the observation just prior to the first therapeutic response (even if it was the predose value).
The systemic pharmacokinetics of F and I were also evaluated as a secondary endpoint, and determined by measuring plasma levels and urinary excretion of both drugs following delivery in 34 patients from two study centres. Plasma concentrations of F or I were measured in blood sampled pre-dose and at 3, 10, 59 and 119 min after inhalation. Urinary excretion of both drugs was evaluated by collecting urine samples pre-dose and in the time intervals 0–0.5 and 0.5–6 h after inhalation of the test drug. The concentrations of F and I were measured using a radioimmunoassay and radioreceptor assay, respectively. The urinary excretion data were pooled from 0–6 h. The limits of quantification of these assays were 20 pg·mL−1 for F in plasma and urine, and 50 and 200 pg·mL−1 for I in plasma and urine, respectively. The radioreceptor assay was not sensitive enough to measure the concentration of I in plasma up to the nominal dose of 40 µg with both devices. Consequently, the pharmacokinetic evaluation of I is confined to the urinary data, pooled from 0–6 h.
Physical examinations and laboratory safety screens were carried out upon admission, and on the last test day. Cardiac frequency and blood pressure (BP) were monitored and recorded before testing on all test days. A 12-lead electrocardiography (ECG) recording was made at screening and on the last test day (≥5 h after inhalation). All adverse events were recorded by the investigator with particular attention given to cough, wheezing and paradoxical bronchoconstriction. Paradoxical bronchoconstriction was defined as a ≥15% fall in FEV1 below baseline at or before 5, 15 and 30 min following inhalation of the test drug. Clinically significant changes in vital signs were defined as follows: 1) systolic BP decrease: below 100 mmHg with a decrease of more than 20 mmHg below baseline; 2) systolic BP increase: above 150 mmHg with an increase of 25 mmHg above baseline; 3) diastolic BP decrease: below 60 mmHg with a decrease of more than 10 mmHg below baseline; 4) diastolic BP increase: above 90 mmHg with an increase of more than 10 mmHg above baseline; 5) cardiac frequency decrease: below 50 beats·min−1 with a decrease of more than 10 beats·min−1 below baseline; and 6) cardiac freqency increase: above 100 beats·min−1 with an increase of more than 20 beats·min−1 above baseline.
Statistical analysis
The primary hypothesis of interest was to demonstrate a dose-response relationship over the dose range studied, based on AUC0–6 h. The expected difference inAUC0–6 h between the lowest and highest dose administered via Respimat® was 0.15 L. Allowing for a balanced incomplete 5-period design with 8 treatments, a sample size of 40 patients was required to detect this treatment difference at a two-sided 5% level of significance with 90% power. The primary efficacy analysis was performed on the per-protocol data set including patients with at least predose and one post-dose FEV1 on ≥2 test days, excluding patients with major protocol violations. The intent-to-treat data set, including all patients who had satisfactory pre- and post-dose data on ≥2 test days was used to confirm the results of the primary analysis on the per-protocol data set. Least square means were obtained using analysis of variance (ANOVA) suitable for cross-over studies. The factors included in the ANOVA were centre, patient within centre, period and treatment. ANOVA was used to compare the highest and lowest doses of Respimat®. Therapeutic equivalence between MDI and Respimat® was addressed by calculating the 90% confidence intervals for the adjusted mean difference between each dose of Respimat® and MDI, accompanied by a test of whether the difference between the pairs of treatments are likely to be as much as 0.15 L. More than one dose from Respimat® was expected to be therapeutically equivalent to the MDI and with only 40 patients, the estimates of the means were expected to be imprecise. Therefore, a log dose-response curve was fitted visually and used to select the point on the Respimat® log dose-response curve which falls closest to the standard dose of MDI. Secondary analyses were performed on the per-protocol data set to compare each Respimat® dose and placebo. All analyses were also repeated for the secondary endpoint peak FEV1 increase. The least square means for FEV1 and FVC at each time point were obtained by performing a separate ANOVA at each time point using the model described above. The least square means were plotted against time for each treatment. The numbers of patients with adverse events were tabulated by treatment. Adjusted mean changes in blood pressure and pulse rate from pre-dose were analysed using ANOVA at each time point as described for spirometry. The statistical analyses were performed using the Statistical Analysis System (SAS) (version 6.08, SAS Institute Inc., Cary, NC, USA).
Results
Efficacy
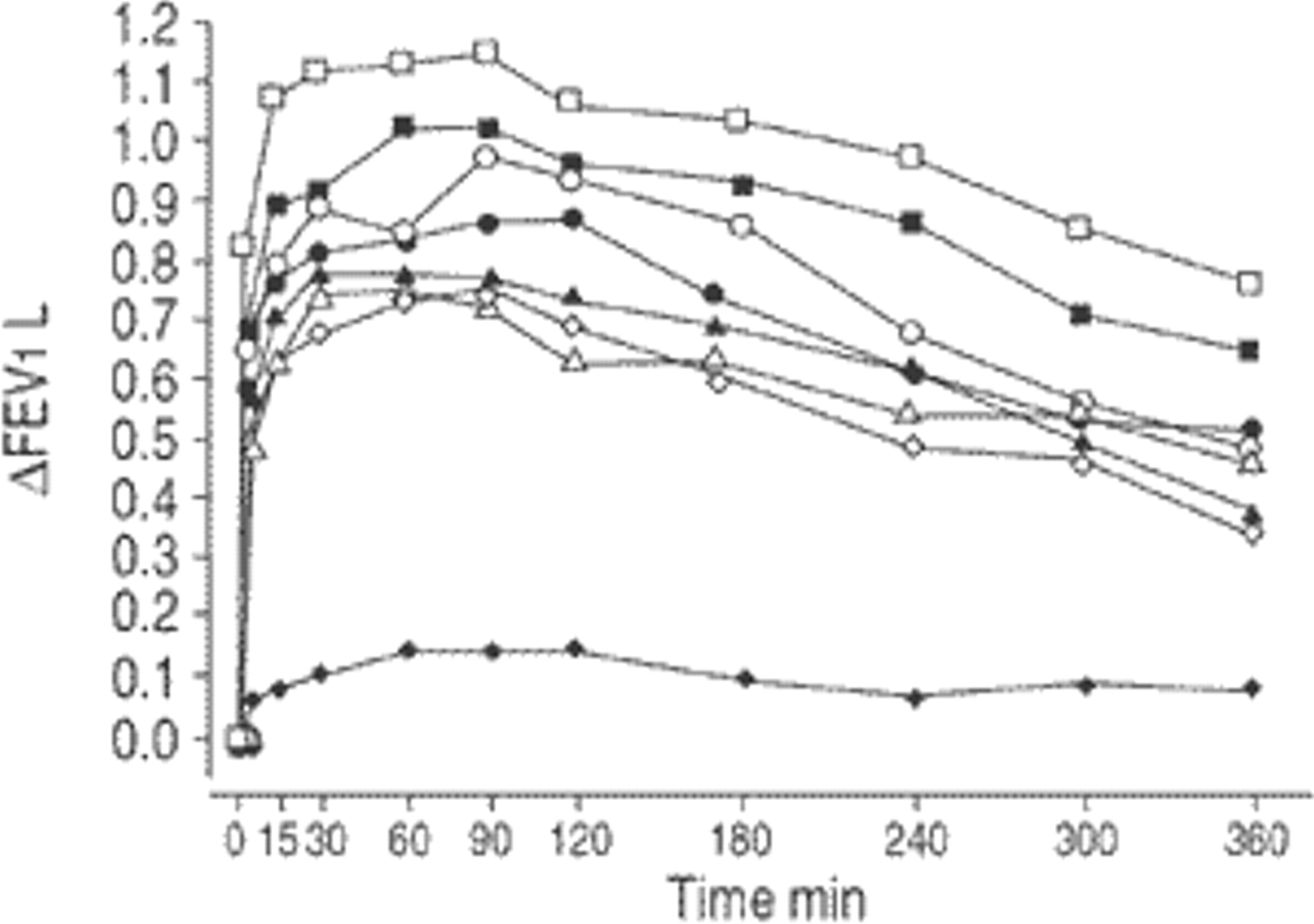
All F/I treatments produced significantly greater increases in FEV1 than placebo (p=0.0001). The adiusted mean time-response curves show the bronchodilator efficacy of all drug treatments across the dose range studied (fig. 1⇓). Adjusted AUC0–6 h, following F/I doses of 12.5/5, 25/10, 50/20, 100/40 and 200/80 µg administered via Respimat® were 0.60, 0.73, 0.79, 0.90 and 1.03 L, respectively, compared with 0.14 L after placebo Respimat® administration. The adjusted AUC0–6 h showed a log-linear dose-response relationship across the dose range administered by Respimat® (fig. 2⇓). A similar relationship was not detected for the two doses of F/I administered by MDI; AUC0–6 h wasslightly higher for the lower dose, at 0.67 L compared with 0.64 L for the higher dose.
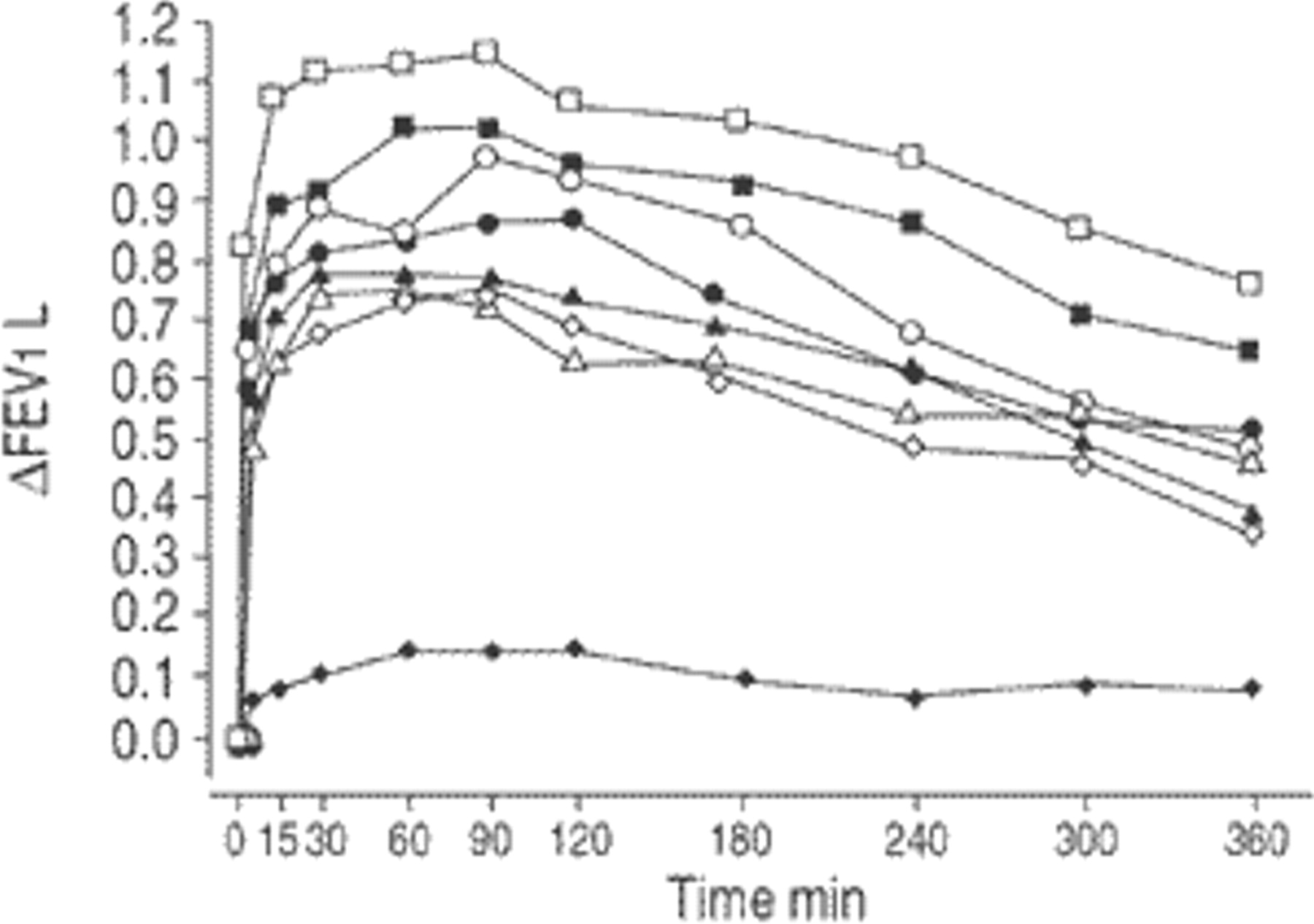
Change in mean forced expiratory volume in one second (ΔFEV1) from pre-dose after inhalation of fenoterol and ipratropium administered by Respimat® (RMT) or metered dose inhaler (MDU) (per-protocol dataset). ⧫: RMT placebo; •: RMT 25/10 µg; ▪: RMT 100/40 µg; ▴: MDI 50/20 µg; ◊: RMT 12.5/5 µg; ○: RMT 50/20 µg; □: 200/80 µg; ▵: MDI 100/40 µg.

Dose-response curve of mean change in forced expiratory volume in one second (AUC0–6 h) from pre-dose. □: Respimat®; ▪: MDI (metered dose inhaler).
A comparison of AUC0–6 h between the lowest and highest doses of F/I (12.5/5 and 200/80 µg) demonstrated that bronchodilation was significantly greater for the higher dose (p=0.0001: treatment difference 0.42 L; 95% confidence interval 0.25–0.59 L). Comparisons between devices for AUC0–6 h failed to demonstrate therapeutic equivalence between any of the F/I doses delivered by Respimat® and the conventional MDI dose (100/40 µg). However, AUC0–6 h values for F/I doses of 12.5/5 and 25/10 µg delivered by Respimat® were closest and slightly superior to that of the 100/40 µg dose via MDI, respectively (fig. 3⇓).

{kind=link}
{kind=link}
{kind=link}
Therapeutic equivalence comparisons for average forced expiratory volume in one second (AUC0–6 h): 90% confidence intervals and p-values for equivalence tests between fenoterol/ipratropium (F/I) via Respimat® and 100/40 µg F/I via metered dose inhaler. ○: adjusted mean differences.
Evaluation of the data for the secondary endpoints in this study produced similar results, with a log-linear dose-response relationship across the range of F/I doses administered by Respimat®, for the adjusted mean changes in peak FEV1 from predose for all eight treatments. Again, no clear dose relationship was evident between the two F/I doses given via MDI.
The median time to onset of the therapeutic response with each active treatment ranged from 2.3–4.6 min. Moreover, the median duration of the response exceeded the 6 h observation period for all treatments except for the 12.5/5 µg dose delivered by Respimat® and the 100/40 µg dose via MDI. The median time taken to reach peak FEV1 was 75–120 min for all active treatments.
The efficacy results for the primary endpoint based on the per-protocol data were confirmed in corresponding analyses for the intent-to-treat patients (n=50, comprising the 47 per-protocol patients and three patients with missing data or protocol violations).
Pharmacokinetics
The plasma concentration and urinary excretion of F are summarised in tables 2, 3 and 4⇓⇓⇓. Administration by Respimat® produced higher plasma values when compared with the same doses delivered by MDI. Excretion of F for equivalent doses also looked higher for Respimat® 100 µg and 50 µg, indicating a trend towards higher doses. Since the plasma concentration of I could not be detected by the radioreceptor assay, only the urinary excretion data are shown (tables 5 and 6⇓⇓). Following delivery of single doses of 20 or 40 µg of I the amounts of drug excretion were significantly higher for Respimat® than the MDI. Overall, the pharmacokinetic studies indicated that the systemic exposure to F and I were proportional to the dose of drug inhaled. Furthermore, there was a two-fold greater systemic availability of both drugs following inhalation via Respimat® compared with the MDI.
Variability of plasma area under curve (AUC) and cumulative urinary excretion of fenoterol
Comparison of adjusted geometric means of area under curve (AUC) data by pairwise t-test
Comparison of adjusted geometric means of urinary excretion data by pairwise t-test
Adjusted geometric means, 95% confidence intervals (CI)
Comparison of adjusted geometric means of urinary excretion data of ipratropium bromide by pairwise t-test
Safety
In general, the treatments were safe and well tolerated. A total of 33 adverse events were reported during the study (table 7⇓). No adverse events were reported with the lowest F/I dose of 12.5/5 µg. The numbers of patients with adverse events were similar following placebo (5/30 patients exposed) and the highest F/I dose via Respimat® (5/30). The incidence of adverse events in the other treatment groups was low and without a clear dose-relationship. No adverse event was seen in more than two patients on any treatment day. Paradoxical bronchoconstriction was experienced by 2 of the 29 patients in the placebo group. These findings were, however, nonsymptomatic and were not recorded as adverse events.
Total drug exposure and adverse events with each treatment
There was one serious event: an asthma exacerbation due to a respiratory tract infection which required overnight hospitalization and was judged to be unrelated to the test drug. The patient recovered and was able to complete the trial as planned. Two adverse events led to withdrawal. One patient withdrew from the study 1–2 weeks after test day 2 due to symptoms of cold, fever and bronchitis. Another patient withdrew from the study on the first test day following drug inhalation (200/80 µg F/I by Respimat®) after experiencing a syncope during blood sampling, attributed to neurovegetative dystonia.
Comparisons between physical examinations, ECG recordings and laboratory screening tests, carried out prior to admission to the study and after completion, showed no differences. The number of patients with clinically significant changes in vital signs was low and balanced across the eight treatments (table 8⇓).
Number of patients with clinically significant changes in vital signs for each treatment
Discussion
In this study, all F/I treatments produced clinically relevant improvements in bronchodilatory efficacy, including a combined F/I dose of 12.5/5 µg via Respimat®. In addition, a log-linear dose-response relationship was obtained across the range of F/I doses administered by Respimat® for the primary endpoint, AUC0–6 h. No dose-response relationship was observed for the F/I doses given by MDI.
Comparison of AUC0–6 h values failed to demonstrate therapeutic equivalence between the MDI and any of the F/I doses administered by Respimat®, due to a substantially higher than expected variability. This may be related to the wide range of responses observed, which is associated with the relatively high level of airway reversibility in this group of patients. Although therapeutic equivalence could not be demonstrated statistically, the responses to F/I doses of 12.5/5 and 25/10 µg administered via Respimat® were closest or slightly superior to that for the F/I dose of 100/40 µg delivered via MDI.
In agreement with results for the AUC0–6 h increase, the dose-response curves of peak increase of FEV1, also indicated log-linearity across the range of F/I doses administered by Respimat® and no clear dose-response relationship between MDI doses.
All F/I treatments showed a rapid onset of effect (medians ranged from 2.3–4.6 min), and extended duration of bronchodilation (over 6 h), with a median time to peak effect ranging from 75–120 min. The pharmacodynamics in the study agree well with those of other studies of F/I administered via MDI. For example, Rammeloo et al. 29 showed an onset of response within 10 min, peak effect after 1 h, and duration of response over 6 h.
Pharmacokinetic analyses demonstrated systemic availabilities of F and I following administration via Respimat®, based on adjusted mean AUC values in plasma and cumulative 0–6 h excretion in urine, were at least double those obtained following administration by MDI. However, as others have emphasized, pharmacokinetic results cannot be directly translated into pharmacodynamic responses 30. The absence of close correlation is supported by the observation of high intersubject variability in spirometric data.
All treatments of F/I were well tolerated whether administered by Respimat® or by the conventional MDI. There was no indication of clinically relevant changes in cardiac frequency or systolic and diastolic blood pressure, with any of the eight study treatments. In addition, there was no worsening in ECG or physical examination compared to baseline, in any of the patients. Overall, the incidence of adverse events throughout this study was generally low. There was a slightly higher incidence of typical systemic beta-adrenergic reactions of nervousness and tremor in the highest Respimat® dosage group, reflecting the higher systemic drug exposure.
The superior device performance of Respimat® when compared to a conventional MDI has also been reported by Maesen et al. 31. In a dose-ranging study, doses of F given via Respimat® or MDI were compared in 61 asthmatic patients. F doses of 12.5 and 25 µg administered by Respimat® were therapeutically equivalent to a 100 µg dose delivered via MDI.
In conclusion, the results of the present study show that fenoterol hydrobromide and ipratropium bromide delivery by Respimat® is as safe and as efficacious in patients with stable asthma, compared with a pressurised metered dose inhaler, at considerably lower doses. Further studies are required to confirm the efficacy and safety of fenoterol hydrobromide and ipratropium bromide delivery by Respimat® in the long-term treatment of patients with asthma and chronic obstructive pulmonary diseases.
- Received July 6, 1999.
- Accepted September 15, 2000.
- © ERS Journals Ltd
References









