Article Text

Abstract
Background Exposure to cigarette smoke (CS) is associated with increased risk of pneumococcal infection. The mechanism for this association is unknown. We recently reported that the particulate matter from urban air simulates platelet-activating factor receptor (PAFR)-dependent adhesion of pneumococci to airway cells. We therefore sought to determine whether CS stimulates pneumococcal adhesion to airway cells.
Methods Human alveolar (A549), bronchial (BEAS2-B), and primary bronchial epithelial cells (HBEpC) were exposed to CS extract (CSE), and adhesion of Streptococcus pneumoniae determined. The role of PAFR in mediating adhesion was determined using a blocker (CV-3988). PAFR transcript level was assessed by quantitative real-time PCR, and PAFR expression by flow cytometry. Lung PAFR transcript level was assessed in mice exposed to CS, and bronchial epithelial PAFR expression assessed in active-smokers by immunostaining.
Results In A549 cells, CSE 1% increased pneumococcal adhesion (p<0.05 vs control), PAFR transcript level (p<0.01), and PAFR expression (p<0.01). Pneumococcal adhesion to A549 cells was attenuated by CV-3988 (p<0.001). CSE 1% stimulated pneumococcal adhesion to BEAS2-B cells and HBEpC (p<0.01 vs control). CSE 1% increased PAFR expression in BEAS2-B (p<0.01), and in HBEpC (p<0.05). Lung PAFR transcript level was increased in mice exposed to CS in vivo (p<0.05 vs room air). Active smokers (n=16) had an increased percentage of bronchial epithelium with PAFR-positive cells (p<0.05 vs never smokers, n=11).
Conclusion CSE stimulates PAFR-dependent pneumococcal adhesion to lower airway epithelial cells. We found evidence that CS increases bronchial PAFR in vivo.
- Cigarette smoke
- platelet-activating factor receptor
- Streptococcus pneumoniae
- adhesion
- airway epithelial cells
- asthma
- macrophage biology
- paediatric asthma
- paediatric lung disease
- paediatric physician
- asthma epidemiology
- asthma guidelines
- asthma mechanisms
- asthma pharmacology
- bronchoscopy
- COPD pathology
- COPD pharmacology
- airway epithelium
- COPD exacerbations
- emphysema
- cystic fibrosis
- innate immunity
- respiratory infection
- aspergillus lung disease
- assisted ventilation
- asthma genetics
- bacterial infection
- pneumonia
Statistics from Altmetric.com
- Cigarette smoke
- platelet-activating factor receptor
- Streptococcus pneumoniae
- adhesion
- airway epithelial cells
- asthma
- macrophage biology
- paediatric asthma
- paediatric lung disease
- paediatric physician
- asthma epidemiology
- asthma guidelines
- asthma mechanisms
- asthma pharmacology
- bronchoscopy
- COPD pathology
- COPD pharmacology
- airway epithelium
- COPD exacerbations
- emphysema
- cystic fibrosis
- innate immunity
- respiratory infection
- aspergillus lung disease
- assisted ventilation
- asthma genetics
- bacterial infection
- pneumonia
Key messages
What is the key question?
-
Does exposure to cigarette smoke increase the adhesion of pneumococci to lower airway cells?
What is the bottom line?
-
Cigarette smoke extract stimulates platelet-activating factor receptor (PAFR)-dependent pneumococcal adhesion to lower airway cells. Increased PAFR was found in mice exposed to cigarette smoke, and in active smokers.
Why read on?
-
We have defined a mechanism underlying the epidemiological association between exposure to tobacco smoke and pneumococcal pneumonia.
Background
Epidemiological studies suggest that exposure to cigarette smoke (CS) is a major risk factor for infection with Streptococcus pneumoniae. Nuorti et al 1 reported that in active smokers, the adjusted risk for invasive pneumococcal disease is 4.1 (95% CI 2.4 to 7.3), and for adults exposed to environmental tobacco smoke (ETS) the adjusted risk is 2.5 (95% CI 1.2 to 5.1). In young children, Suzuki et al 2 reported that exposure to ETS is associated with hospital admissions with pneumonia (adjusted OR 1.55, 95% CI 1.25 to 1.92), and estimated that 28% of pneumonia admissions in Vietnamese children are attributable to ETS. Since S pneumoniae is the major cause of pneumonia,3 these data suggest that risk of pneumococcal pneumonia is increased by active smoking and exposure to ETS. Biological plausibility for this association is provided by a study that found chronic exposure of mice to CS followed by pneumococcal infection increases morbidity and the amount of colony forming unit (CFU) counts of pneumococci in the lung.4
To date, the mechanism whereby CS increases vulnerability to pneumococcal pneumonia is unclear. A prerequisite step for the development of pneumococcal infection is the adhesion of pathogenic bacteria to lower airway epithelial cells.3 The capacity of pneumococci to adhere to airway cells is enhanced by acid,5 respiratory viral infection,6 and interleukin (IL)-1α.7 Adhesion of virulent strains of S pneumoniae to bronchial and alveolar airway epithelial cells is mediated, in part, by phosphorylcholine in the bacterial cell wall binding to the receptor for platelet activating factor receptor (PAFR) on host cells.5–7 During the normal processes of PAFR internalisation, pneumococci bound to the receptor are transported into cells—that is, pneumococci use PAFR as a Trojan horse.7 We previously reported that exposure of airway cells to urban air particulate matter (PM) in vitro increases the adhesion of S pneumoniae via the PAFR.8 Since both CS and urban PM share pollutants, for example, carbonaceous PM, we hypothesised that CS upregulates pneumococcal adhesion to lower airway cells via PAFR. In this study, we sought to assess the effect of cigarette smoke extract (CSE) on pneumococcal adhesion in a human lower airway cells. Using stored tissue from mouse and human studies, we then sought evidence that CS exposure increases PAFR expression in vivo.
Methods
A549, BEAS2-B, primary bronchial epithelial cells and S pneumoniae
A549, a human type II pneumocyte cell line, was obtained from Sigma Aldrich (Poole, UK). BEAS2-B cells were kindly donated by Dr Nicolas Mercardo (National Heart and Lung Institute, Imperial College London, UK). Human primary bronchial epithelial cells (HBEpC) were purchased from Promocell (Heidelberg, Germany). Cells were maintained in Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum and 1% L-glutamine/penicillin-streptomycin (Lonza, Basel, Switzerland). Passage number was <10 for bronchial epithelial cells, and <15 for A549 cells. We aimed to fully define the effect of CSE on pneumococcal adhesion and PAFR in A549 cells, then replicate key findings in BEAS2-B and primary human bronchial epithelial cells. The virulent type 2 S pneumoniae encapsulated strain D39 (NCTC 7466) was purchased from the National Collection of Type Cultures (Central Public Health Laboratory, London, UK) and grown to mid-log phase (optical density 600=0.5) prior to adding to A549 and primary bronchial cells at 200 μl, 2×108 mid-logarithmic phase, and 100 μl for BEAS2-B cells.
Generation of CSE
CSE in filter material was prepared at the Institute of Clinical Science (Lund, Sweden), as previously described.9 Briefly, three cigarettes (0.8 mg nicotine per cigarette; Marlboro; Philip Morris USA, Pittsburgh, Pennsylvania, USA) were ‘smoked’ by a water aspirator, and CS aspirated through a cotton wool filter. CSE (100%) was obtained by vortexing the cotton wool filter in 2 ml dimethyl sulfoxide (DMSO).9 DMSO was stored at −20°C as 100% stock solution. Stock solution was then diluted in medium and used for all experiments at ≤1%. We previously determined that the nicotine content of CSE was 0.1–0.15 mg/ml.9 CSE was diluted in medium and used in experiments at ≤1%. DMSO ≤1% did not alter pneumococcal adhesion.
Cell viability in A549 cells exposed to CSE in vitro
The effect of CSE on cell viability was assessed in A549 cells. Lactate dehydrogenase (LDH) release, a marker of cell membrane integrity, was done according to the manufacturer's protocol (Sigma Aldrich, Gillingham, UK). The assay included LDH released after total cell lysis (‘total’ LDH control). Optical density of LDH was assessed by spectroscopy. Cell cytotoxicity was assessed by the MTT (3-(4,-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium) assay.10 This assay assesses the conversion of the MTT reagent to formazan which then accumulates in healthy cells. Briefly, 20 μl MTT (5 mg/ml, Sigma Aldrich) in phosphate-buffered saline was added to A549 cells, and incubated for 1 h. Medium was removed and 100 μl of DMSO (Sigma Aldrich) added for 30 min. Absorbance (optical density units) was assessed at 550 nm. In the MTT assay, cytotoxicity reduces the optical density.
PAFR transcript level in airway cells exposed to CSE in vitro
Transcript level of PAFR was assessed in A549 cells by quantitative real-time PCR. For A549 cells, 2×105 cells were harvested for each sample and RNA was prepared using the RNeasy mini kit (Qiagen, Crawley, UK). Random hexamers (Qiagen) and M-MLV reverse transcriptase (Invitrogen, Paisley, UK) were used to synthesise first strand cDNA according to manufacturer's instructions. mRNA transcript level of PAFR and the house-keeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were determined by quantitative real-time PCR using an ABI 7500 real-time PCR system (Applied Biosystems, Warrington, UK) with TaqMan primer and probe sets HS00265399_S1 (PAFR), and Hs99999905_m1 (GAPDH) respectively. mRNA transcript level was normalised to GAPDH, and the relative change in expression between samples calculated using the comparative CT method according to the manufacturer's instructions (Applied Biosystems). Data are representative of two separate experiments with six replicates.
PAFR expression in airway cells exposed to CSE in vitro
PAFR expression in A549, BEAS2-B and HBEpC was determined by flow cytometry (details in online supplement). Specific PAFR median florescence intensity was determined for each concentration of CSE by subtracting the median florescence intensity of the isotypic control monoclonal antibody (mAb) from the median florescence intensity of the PAFR mAb. Data are expressed as fold-change in PAFR over control, and are from four experiments done at different times for A549 and three experiments done at different times for BEAS2-B cells. Due to limited cell number, PAFR expression in HBEpC is from one experiment.
Pneumococcal adhesion to airway cells exposed to CSE in vitro
Adhesion of pneumococci to airway cells in vitro was done using a standard bacterial adhesion assay.7 ,8 ,11 The adhesion assay reflects adherent and intracellular bacteria. Briefly, A549 cells and HBEpC at 2×105/ml were seeded in 24-well plates (Costar, Sigma Aldrich). CSE up to 1% was added to cell monolayers and incubated for 4 h at 37°C. CSE was removed by washing twice with Dulbecco's Modified Eagle Medium (Lonza) and pneumococci added and incubated for 2 h.5 Cell monolayers were washed five times and cells removed from the tissue culture plate by trypsin-EDTA, and lysed with ice-cold sterile distilled water for 10 min. Lysates were plated out for colony forming units (CFU) /mL.12 For BEAS2-B cells, the adhesion assay was modified. Briefly, cells were maintained in DMEM and then seeded at 2×105 cells/ml in Promocell Bronchial Epithelial Cell Media (Promocell) for 48 h. The last washing step reduced to three times before plating. The functional relevance of PAFR was assessed by co-incubating cells with the competitive PAFR antagonist CV-398813 (Sigma Aldrich). A stock solution of CV-3988 was prepared at 1 mM in ethanol then diluted in medium to a final concentration of 10 μM—a concentration that inhibits PAF-induced cortisol release from adrenal glands.14 We established that CSE per se does not stimulate pneumococcal proliferation, and that ethanol at the concentration in 10 μM CV-3988 does not affect pneumococcal adhesion. Adhesion data are representative of at least three separate experiments done on different days for A549 and BEAS2-B cells, and two experiments done on different days for HBEpC.
Lung PAFR transcript level in mice exposed to CS in vivo
Six to eight-week-old female C57BL/6 mice (purchased from Charles River Laboratories, Montreal, Canada) were exposed to mainstream CS using a whole body CS exposure system (SIU48, Promech Lab AB, Vintrie, Sweden), as previously reported in detail.15 Briefly, mice (n=5) were exposed to the mainstream CS of 12× 2R4F reference cigarettes with filter removed (Tobacco and Health Research Institute, University of Kentucky, Lexington, Kentucky, USA) over 50 min, twice daily. Control animals (n=5) were exposed to room air only. Following 4 days of CS exposure, lungs were removed and snap frozen in liquid nitrogen. Samples were sent to the UK on dry ice for PAFR mRNA analysis. Unfrozen tissue samples were homogenised and the RNA extracted as described above. First strand cDNA synthesis was carried out using the high-capacity RNA-to-cDNA master mix (Applied Biosystems), according to the manufacturer's instructions. mRNA transcript level of PAFR and house-keeping gene, β2 microglobulin, were determined by quantitative real-time PCR as described above, with TaqMan primer and probe sets Mm02621061_m1 and Mm00437762_m1 respectively. Mouse PAFR mRNA transcript level was normalised to the housekeeping gene β2 microglobulin. McMaster University's Animal Research Ethics Board approved all animal procedures.
Bronchial PAFR expression in adults exposed to CS in vivo
Bronchial biopsies were obtained for active smokers and healthy never smokers as previously described.16 Subjects gave written, informed consent approved by the Human Research Ethics Committee (Tasmania) Network (approval number: H0007017). Subjects with a history suggestive of asthma, other respiratory disorders and uncontrolled comorbidities were excluded. Never smokers had no history of respiratory illness or smoking. For smokers, the inclusion criteria were a minimum 10-pack-year history of cigarette smoking with spirometry within normal limits post bronchodilator (forced expiratory volume in a 2 (FEV1)>80% of predicted, and FEV1/forced vital capacity>70%). Endobronchial biopsies were taken from subsegmental carinae of the right lower lobe of each patient, using alligator forceps (FB-15C; Olympus, Tokyo, Japan). Bronchial biopsies were fixed in 4% neutral buffered formalin for 2 h and subsequently processed into paraffin through graded alcohol and xylene using a Leica ASP 200 tissue processor. Sections from stored specimens were cut at 3 μm from individual paraffin blocks, stained with H&E and morphologically assessed for immunostaining (details in online supplement). Expression of PAFR was measured on randomised and coded slides by an operator blinded to smoking status and expressed as the percentage of epithelium with PAFR-positive cells for each subject.
Statistical analysis
Normally distributed data are summarised as mean (SEM) and analysed using GraphPad Prism version 5.03 (GraphPad Software, La Jolla, California, USA), with analyses done by unpaired t test. For non-normally distributed data, comparisons were done by Mann–Whitney U test. For mRNA transcript number, the mean of raw data of control samples was assigned 100% and the SEM converted to a percentage of the raw value. In human tissue, PAFR mRNA data were normalised to mRNA from the housekeeping gene GAPDH, and in mouse tissue normalised to β2 microglobulin. A p value <0.05 was considered significant.
Results
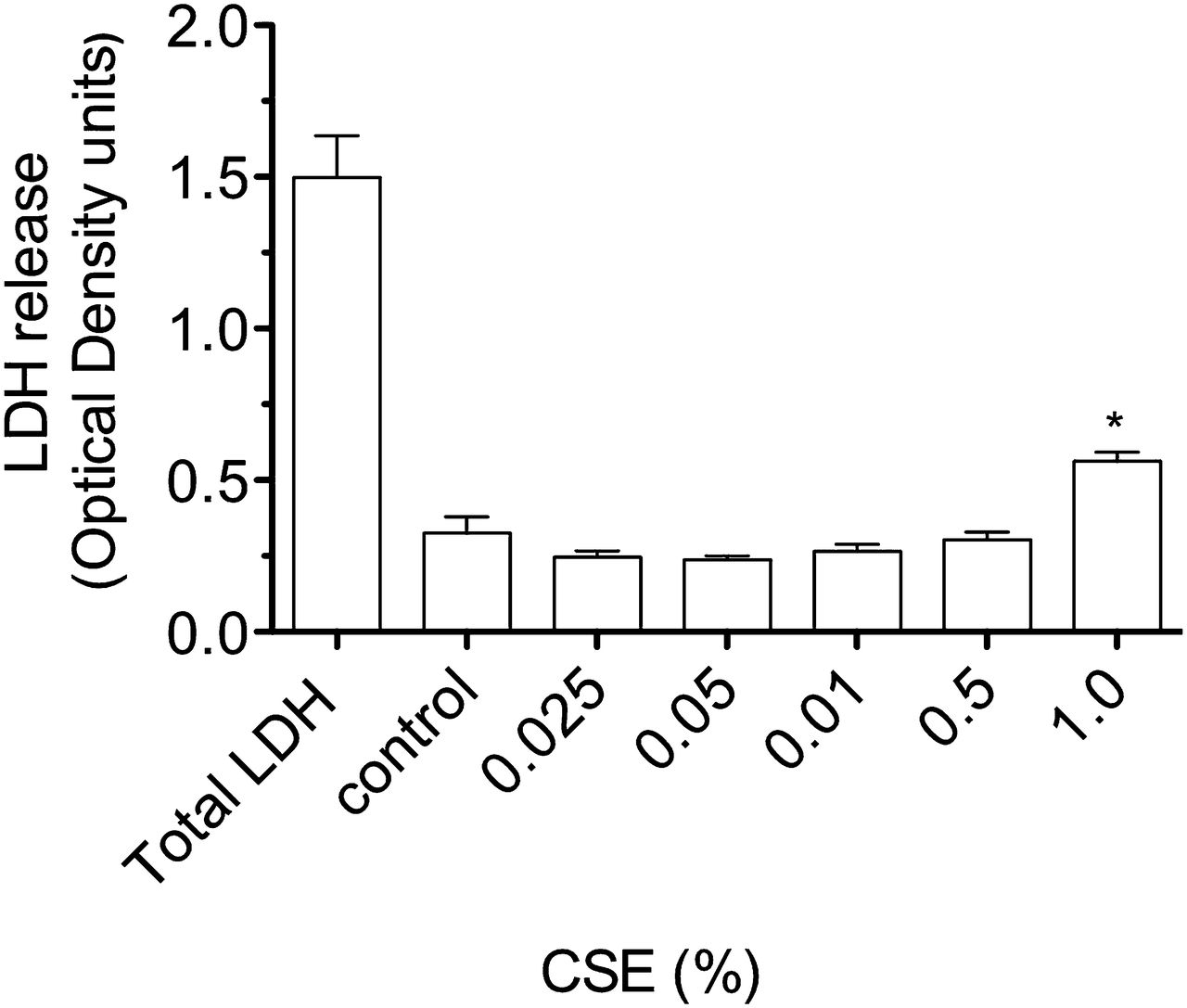
Exposure of airway cells to CSE 1% for 4 h in vitro resulted in cell contraction, cytoplasmic vacuolation, and a tendency for cells to detach from the culture plate on washing (online figures 1 and 2). Brown PM was visible on cells cultured with CSE 1%, but at a relatively low density compared with the concentration of urban PM (50 μg/ml) reported to stimulate pneumococcal adhesion8 (online figure 1). Culture of A549 cells with CSE 1% for 4 h was not cytotoxic as assessed by the MTT assay (figure 1). A moderate increase in LDH release by A549 cells was stimulated by CSE 1% (p<0.01 vs medium control, figure 2).

Conversion of MTT (3-(4,-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium) by A459 cells stimulated with cigarette smoke extract (CSE) for 4 h. Optical density (OD) of the formazin product is detected by spectroscopy at 550 nm. CSE does not attenuate in the ability of cells to convert MTT, indicating an absence of cytotoxicity. CSE increased conversion at 0.5 and 1% (*p<0.05 vs medium control).
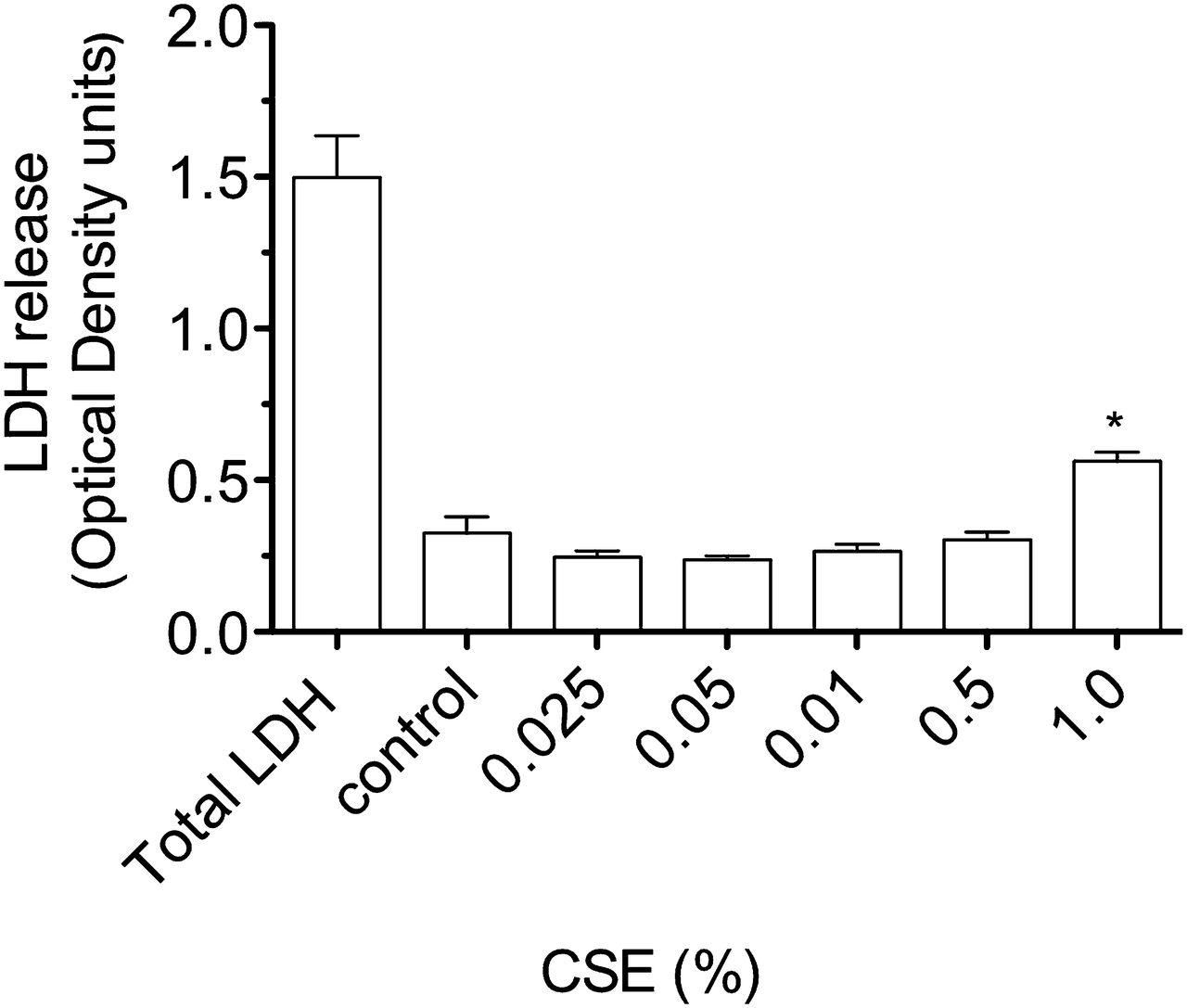
Release of lactate dehydrogenase (LDH) by A549 cells stimulated with cigarette smoke extract (CSE) for 4 h. Compared with LDH release after lysis of all cultured cells (total LDH), there is a small, but significant, increase in LDH by cells stimulated by CSE 1% (*p<0.01 vs medium control).
CSE at 0.5% and 1% increased pneumococcal adhesion to A549 cells (p<0.05 vs control, figure 3A). Co-incubation with a specific PAFR antagonist (CV-3988) attenuated CSE-stimulated adhesion to A549 cells (p<0.001; CSE 0.5%+antagonist vs CSE 0.5%, figure 3B), but had no effect on basal adhesion (data not shown). CSE 1% increased PAFR transcript level (p<0.01 vs control, figure 3C), and PAFR expression in A549 cells (p<0.01 vs control, figure 3D). CSE 1% increased pneumococcal adhesion to BEAS2-B cells and HBEpC (p<0.01 vs control, figure 4A,B), and increased PAFR expression in BEAS2-B cells (1.9±0.07-fold increase, p<0.05 vs control), and HBEpC (3.4±0.8-fold increase vs control).

Effect of cigarette smokes extract (CSE) on A549 cells. (A) Incubation with CSE 0.5% and 1% increases Streptococcus pneumoniae colony-forming units (CFU), indicating increased adhesion (*p<0.05 vs medium control). Data are represented as mean and SEM (n=8, and n=6 respectively). (B) Coincubation with a platelet-activating factor receptor (PAFR) blocker (10 μM CV-3988) attenuates CSE-stimulated adhesion (*p<0.001 vs without PAFR blocker). Dimethyl sulfoxide (DMSO) at 1% does not affect adhesion. Data are represented as mean and SEM (n=8), and are representative of four separate experiments. (C) CSE 1% increases PAFR transcript level (*p<0.01 vs medium control). PAFR was assessed by quantitative real-time PCR and normalised for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data are representative of two experiments with six replicates. (D) CSE 1% increases PAFR expression. Cells were immunostained with either mouse antihuman PAFR, or isotypic control and expression determined by flow cytometry. Data are described as fold change in median florescence intensity (MFI) over medium control, adjusted for the isotypic control. Data are expressed as mean (SEM) fold change, and are from four separate experiments (*p<0.01 vs control).

Effect of cigarette smoke extract (CSE) on the adhesion of Streptococcus Pneumoniae to; (A) BEAS2-B cells, and (B) human primary bronchial epithelial cells (HBEpC). CSE 1% increases adhesion to both cell types (*p<0.05, and **p<0.01 vs control, n≥5). Data are expressed as mean (SEM) and are representative of four separate experiments for BEAS2-B, and two separate experiments for primary bronchial cells. CFU, colony forming unit.
In mice, 4-day exposure to CS increased lung PAFR transcript level (n=5 per group, p<0.05 vs room air, figure 5). In humans, 6/16 active smokers had PAFR-positive bronchial cells (0.05, 0.1, 0.58, 2.12, 2.25, 4.82% of bronchial epithelium with positive cells respectively, figure 6). In contrast, all 11 never smokers had occasional very low levels of PAFR staining that were always below the positive cutoff threshold (active smokers vs never smokers, p<0.05, Mann–Whitney U test). There was no significant difference in sex and age between smokers and non-smokers (Table 1) and no difference in age, pack-years or lung function between the PAFR-positive and PAFR-negative active smokers. Non-specific staining levels were low in both active and never smokers (online figure 3).
Demographics and lung function data for adults undergoing bronchoscopy and bronchial biopsy

Exposure of mice to cigarette smoke for 50 min twice a day for 4 days increases pulmonary platelet-activating factor receptor (PAFR) transcript level (*p<0.05 vs room air control). PAFR was assessed by quantitative real-time PCR and transcript level normalised to β2 microglobulin. Data are from a single in vivo exposure experiment with five animals in each group.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Active smoking and bronchial platelet-activating factor receptor (PAFR) expression. (A) Section of a bronchial biopsy from one of the six active smokers with PAFR-positive cells. Ten active smokers had no cells that reached the positive threshold. PAFR staining is brown and is most pronounced in the apical part of the bronchial epithelium (arrowed). (B) A representative bronchial biopsy from a never smoker showing low PAFR staining that is below the cutoff threshold for positivity. Original magnification ×400. Scale bar = 50 μm. The isotypic control is shown in online figure 3.
Discussion
In this study, we found that in vitro exposure to CSE upregulates the adhesion of pneumococci to lower airway epithelial cells. A major role for PAFR in CSE-stimulated adhesion is suggested by increased PAFR mRNA transcript level and PAFR expression in CSE-exposed A549 cells. Furthermore, we found that adhesion was attenuated by a specific PAFR antagonist (CV-3988). Compatible with previous studies in cytokine-stimulated A549 cells,7 blocking PAFR attenuated stimulated adhesion, but not unstimulated adhesion of pneumococci to A549 cells. The effect of CSE on adhesion is not limited to the most distal epithelial cells, since CSE stimulated adhesion to a bronchial epithelial cell line (BEAS2-B) and to human primary bronchial cells (HBEpC). The clinical relevance of upregulation of PAFR in vivo is suggested by several animal models. For example, interleukin 1 stimulates PAFR in the rabbit airway, which in turn increases vulnerability to pneumococcal pneumonia.7 Similarly, influenza infection increases PAFR in the mouse lung which increases lethality to pneumococcal infection,17 and increased lung PAFR in mice transplanted with sickle-cell bone marrow causes hypersusceptibility to pneumococcal infection.18 Conversely, PAFR–/– mice with reduced PAFR expression in airway cells are resistant to lethal pneumococcal pneumonia.19 In this study, we found evidence that CS upregulates PAFR in vivo, since PAFR transcript level was increased in the lungs of mice exposed to CS, and bronchial epithelial PAFR expression was increased in active smokers.
The mechanism whereby CSE upregulates airway epithelial PAFR is unclear. Cell death was excluded since, compatible with other studies,20 CSE 1% did not attenuate cell function assessed by the MTT assay. CSE did however stimulate a moderate increase in LDH release. Previous studies report that tobacco smoke, via induction of oxidative stress, stimulates bronchial epithelial cell proliferation,21 activates stress-activated protein kinase,22 and disrupts airway cell cytoskeleton proteins.23 In this study, the tendency of CSE-exposed cells to contract, detach from the tissue culture plate, and release LDH is compatible with a response to oxidative stress. Since CSE contains thousands of bioactive chemical compounds—including carbonaceous PM, nicotine, alkaloids, metals, nitrosamines, polyaromatic hydrocarbons, aromatic amines, organic compounds,24 and endotoxin25—the component stimulating pneumococcal adhesion remains unclear. We previously reported that fossil-fuel PM per se, albeit at high density, stimulates pneumococcal adhesion to lower airway cells (online figure 1A to F).8 However, the density of PM on cells exposed to CSE 1% was relatively low (online figure 1A to F). We therefore speculate that the PM and soluble fractions of CSE stimulate adhesion. Evidence for the bioactivity of soluble components of CS is provided by our previous study in vascular and bronchial smooth muscle cells in which CSE, but not a suspension of PM from CS, upregulated immune receptor expression.9 Furthermore, Sohn et al 26 assessed the effect of three soluble CS components (nicotine, benzopyrene and napthylamine) on immune response-related genes in A549 cells, and reported that PAFR was one of only 39/1152 genes upregulated by nicotine.26
Previous studies support the hypothesis that inhalation of CS upregulates pneumococcal adhesion to lower airway cells in vivo. For example, Riise et al 27 reported a non-significant trend for increased adhesion of S pneumoniae to fixed airway cells from smokers with chronic bronchitis and El Ahmer et al 28 reported increased binding of S pneumoniae to buccal epithelial cells from active smokers compared with non-smokers. To date, data on PAFR in the human lung are conflicting. On one hand, Ishizuka et al 5 reported PAFR expression by cultured ciliated tracheal epithelial cells obtained postmortem. On the other hand, Shirasaki et al 29 found no PAFR mRNA in airway epithelial cells from heart–lung transplant donor lungs, and lower levels of PAFR mRNA in whole lung tissue from smokers (n=4) compared with non-smokers (n=8).29 In contrast, we found increased PAFR transcript level in mouse lung after controlled exposure to CS, and evidence of increased PAFR expression in bronchial biopsies from active smokers. The reason why we found PAFR-positive cells in only a subgroup of smokers is unclear. We speculate that immunostaining of stored, fixed tissue is a relatively insensitive technique for assessing PAFR expression. Alternatively PAFR expression may be determined by variables that we did not record, for example, time from last cigarette.
There are limitations to this study. First, we did not model the antioxidant and surfactant-rich layer of epithelial lining fluid that provides protection against inhaled pollutants in vivo.30 Second, there are very likely to be several other factors, apart from adhesion, contributing to CS-induced vulnerability to bacterial infection in vivo. For example, CS attenuates human β-defensin-2 production in airway cells,31 and in murine models reduces complement-mediated phagocytosis of S pneumoniae by airway macrophages,4 and reduces ciliary beat frequency.32 Third, CSE in solution may not fully reflect airway deposition at an air–tissue interface.33 Finally, additional mechanisms may be involved in mediating pneumococcal adhesion in vivo. In upper airway epithelial cells the polymeric immunoglobulin receptor contributes to pneumococcal adhesion,34 although this receptor is not expressed by A549 cells (S. Hammerschmidt, personal communication).
In conclusion, exposure of lower airway epithelial cells to CSE in vitro upregulates PAFR-dependent pneumococcal adhesion. Increased lung PAFR mRNA in mice exposed to CS, and increased PAFR expression in the bronchial epithelium of active smokers, suggests that active and passive inhalation of CS upregulates airway PAFR in vivo.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
-
Funding This work was supported by the Barts and the London Charity, and grants from the Swedish Research Council (number 05958), and the Royal Hobart Hospital Research Foundation, and NHMRC Australia.
-
Competing interests None.
-
Ethics approval Human Research Ethics Committee (Tasmania) Network (approval number: H0007017).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Raw data will be provided on request.