Article Text

Abstract
Dyskeratosis congenita (DC) is a rare inherited multisystem disorder characterised by lesions of the skin and appendages. Bone marrow failure occurs in 80% of patients. The gene for the X-linked form of DC has been identified on Xq28 and designated as DKC1. Pulmonary manifestations have rarely been reported. It is not known whether there is a respiratory disease peculiar to these patients and, if so, whether it is associated with a specific genetic mutation. A 40 year old Egyptian man with pulmonary disease and his symptom free 35 year old brother both presented with mucocutaneous lesions characteristic of DC. In the older brother chest imaging revealed generalised intralobular interstitial thickening and honeycombing. Pulmonary function tests showed a restrictive pattern. Open lung biopsy specimens of lung tissue showed various degrees of fibrosis consistent with usual interstitial pneumonia of chronic idiopathic pulmonary fibrosis. The younger brother was free of pulmonary lesions. Both had a novel missense mutation 5C→T in exon 1 of the DKC1 gene. It is concluded that pulmonary disease in DC may be underestimated, possibly because most patients die at an early age of bone marrow failure. No relationship between genotype and phenotype could be established in the patients studied. The genetic diagnosis of DC is now available, which may enable it to be diagnosed in patients with restrictive pulmonary disease and minimal cutaneous signs.
- dyskeratosis congenita
- restrictive pulmonary disease
- DKC1 gene
- United Arab Emirates
Statistics from Altmetric.com
Dyskeratosis congenita (DC), or Zinsser-Engman-Cole syndrome, is a rare inherited multisystem disorder characterised by reticulate skin pigmentation, nail dystrophy, hypotrichosis, and mucosal leukoplakia. A number of non-cutaneous features have also been reported including epiphora, early dental loss, skeletal, gastrointestinal, and urinary abnormalities. Bone marrow failure occurs in 80% of cases and there is a predisposition to malignancy, especially of the gastrointestinal tract. Pulmonary manifestations have rarely been reported and are often not well documented; their incidence is possibly underestimated. Most cases of DC are transmitted in an X-linked recessive mode. Some, however, are consistent with autosomal inheritance.1
The gene whose abnormalities are responsible for the X-linked form of DC has been identified and designated as DKC1.2 This gene maps to Xq28 and encodes a protein of the dyskerin family that is predictably involved in both the cell cycle and nucleolar function.3 ,4
We report the case of two brothers with X-linked DC in whom a novel mutation was found. One brother presented with diffuse interstitial pneumonia of the usual interstitial type while the other was free of pulmonary disease. We also review the literature with a view to establishing the incidence and types of pulmonary disease in DC, and we probe possible genotype-phenotype relationships in the condition.
Case report
A 40 year old Egyptian man was referred in October 1997 to the Chest Clinic at Tawam Hospital with a 7 month history of progressive dyspnoea on exertion and minimal cough. He had smoked 20 cigarettes a day from adolescence until 1990 when he stopped smoking. He was working in an office without exposure to dust and chemicals. Since the age of 10 he had noticed skin changes that had been worsening with time.
The patient's condition at examination was good. His height was 165 cm, weight 77 kg, the pulse was regular at 70/min, blood pressure 150/90 mm Hg, and temperature 36.8°C. Chest examination revealed poor expansion and diffuse bilateral lower zone crackles. Cardiovascular, abdominal, and neurological examinations were normal.
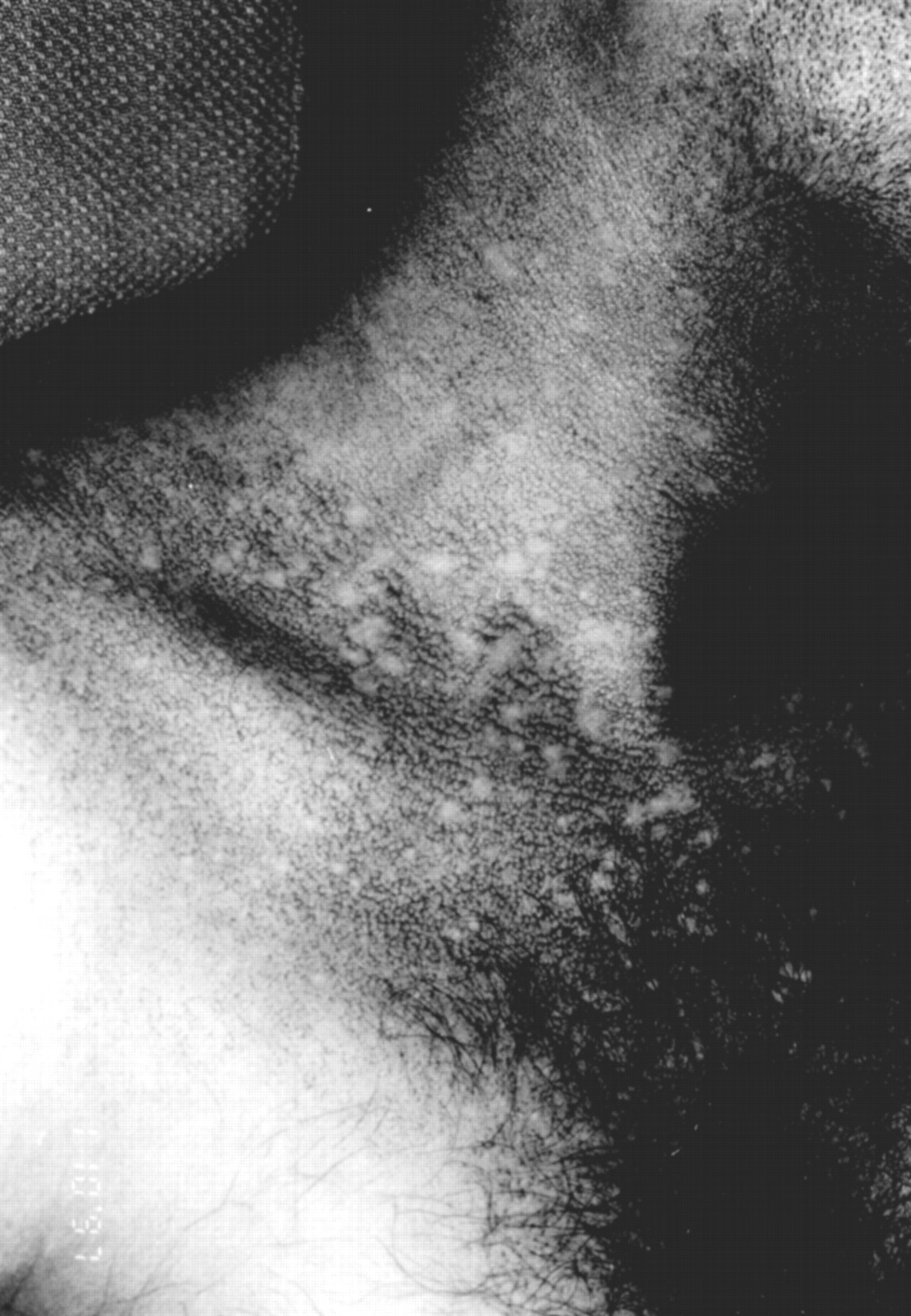
Skin examination revealed a widespread reticulate pigmentation on the neck and upper chest where “confetti” spots of spared skin were present (fig 1). The upper eyelids, cheeks (where telangiectases were also present), auricles, extensor aspects of the upper limbs, palms, lower abdomen, scrotum, and thighs were also involved. In addition, there was a marked leukoplakia of the tongue. The fingernails were over-curved, evoking a mild clubbing, and the toenails were dystrophic with partial atrophy of the smaller ones. The hair in the outer halves of the eyebrows was sparse and there was diffuse, non-scarring alopecia of the scalp. There were breaks in the dermatoglyphics and in the epidermal ridge pattern or the palms. His teeth, testes, hearing, and sight were normal.
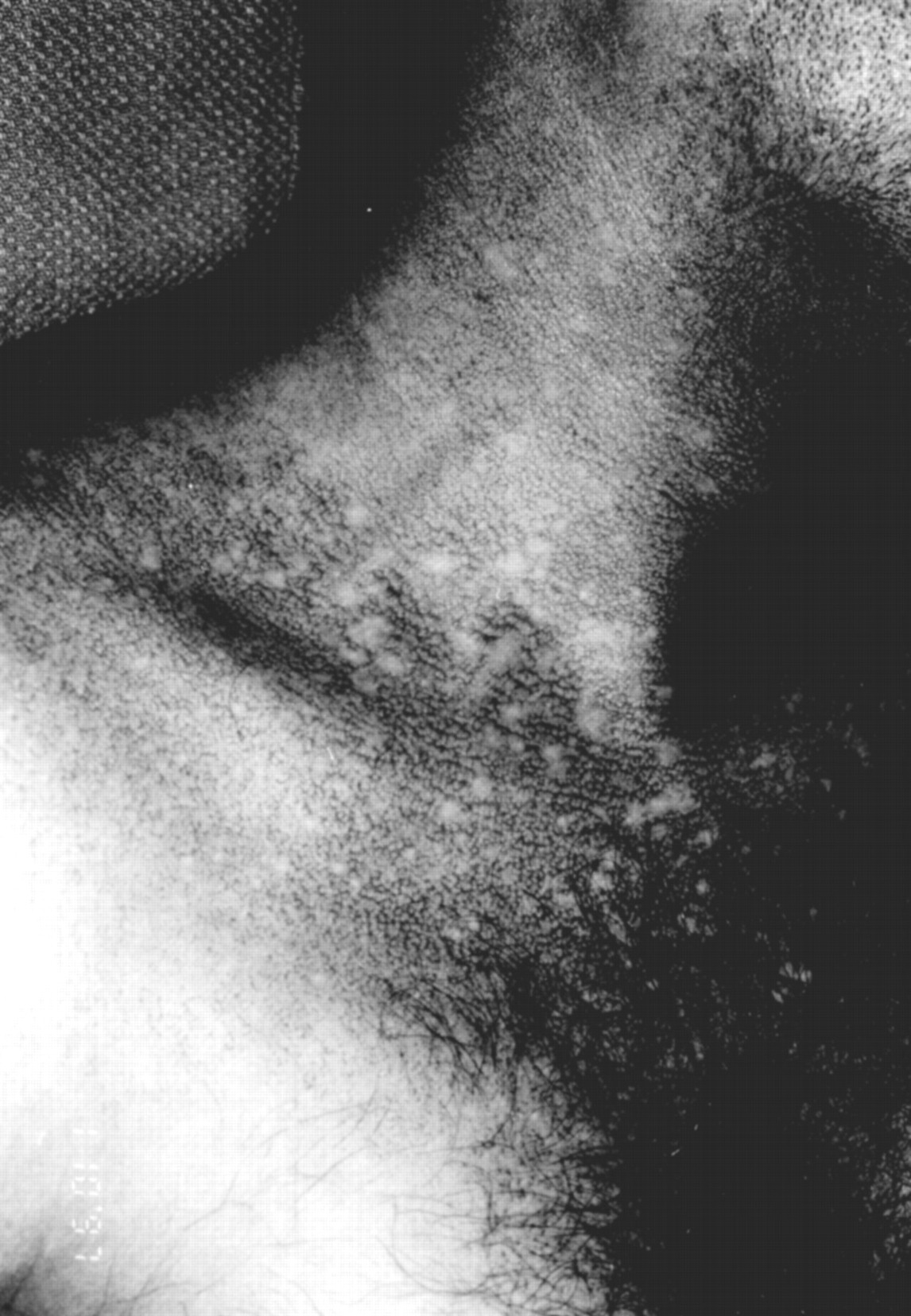
Neck and upper chest: reticulate pigmentation with “confetti” spots of spare skin.
IMAGING
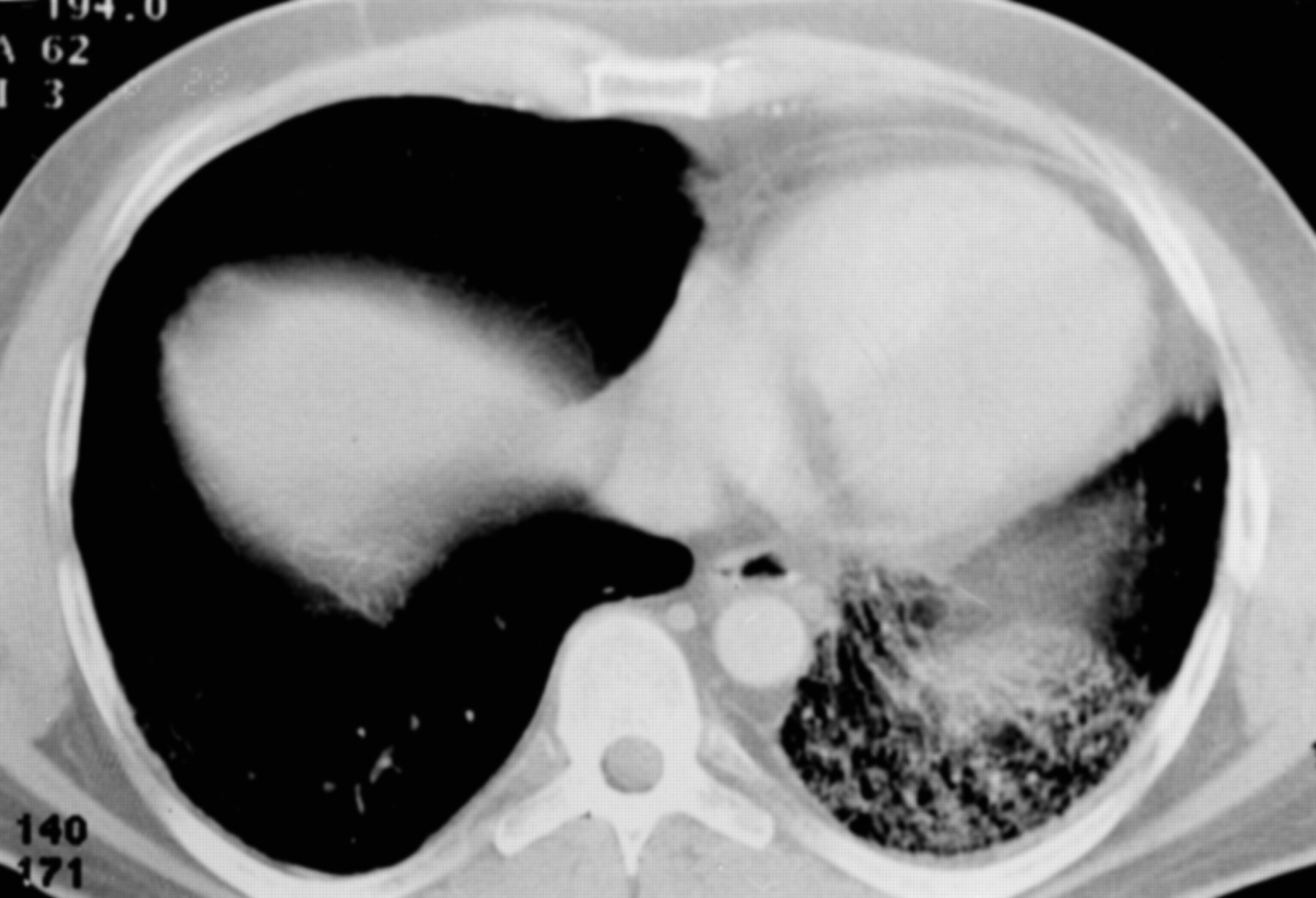
Chest radiographs and computed tomographic (CT) scans revealed extensive overall reduction of the lung volume on the left side (fig2). There was extensive left sided apical pleural thickening, minor pleural thickening in the lateral aspect of the right lung, and moderate pleural thickening in both lung bases. Intralobular interstitial thickening and honeycombing were most marked in the posterior aspect of the left lower lobe and milder in the peripheral and subpleural areas of the left middle and upper lung zones. In the right lower lobe posterior segment there were diffuse ground glass opacities and, in the rest of the right lung, there were patchy interstitial infiltrates and minor areas of honeycombing in the subpleural area. Oesophageal transit time was normal.

Chest CT scan of lower lobes showing bilateral interstitial thickening more marked on the left side, mostly in the posterior aspect of the lobe.
INVESTIGATIONS
Peripheral blood count showed a normal red blood cell count and a low platelet count (121 × 109/l (normal 150–400 × 109/l). The white blood cell count was 4.0 × 109/l (normal 4.0–11.0 × 109/l) and the lymphocyte count was 1.2 × 109/l (normal 1.2–3.4 × 109/l) with CD4 20% (normal 28.5–60.5%) and CD8 64% (normal 11.1–38.4%); the CD4/CD8 ratio was 0.31 (normal >1). Routine biochemical and liver function tests were normal. The cholesterol level was 6.53 mmol/l (normal 3.10–5.60 mmol/l) and HDL and triglyceride levels were normal. Serum protein electrophoresis was normal without monoclonal bands. Serological tests for hepatitis B and C, HIV, rheumatoid factor, anti-nuclear and anti-DNA antibodies were negative. IgM was low (0.38 g/l; normal 0.60–2.50 g/l); IgA and IgG were normal. Urinalysis was normal.
Arterial blood gas tensions (room air) were as follows: pH 7.40 (normal 7.36–7.44), Paco 2 5.15 kPa (normal 4.8–5.9<thin), Pao 2 8.16 kPa (normal ⩾10), and HCO3 – 23.9 mmol/l (normal 24–30).
Pulmonary function tests revealed a restrictive pattern. Forced vital capacity (FVC) was 1.69 l (mean (SD) predicted value 4.5 (1.5) l), forced expiratory volume in 1 second (FEV1) 1.43 l (predicted 3.5 (1) l), FEV1/FVC 85% (predicted 77%), total lung capacity (TLC) 2.85 l (predicted 6.0 (1.5) l), and residual volume (RV) 1.16 l (predicted 1.8 (0.8) l).
Sputum cultures grew Haemophilus influenzaeand were negative for fungal and acid-fast bacilli. Fibreoptic bronchoscopy was non-contributory and bronchoalveolar lavage revealed only a slightly raised percentage of lymphocytes.Pneumocystis carinii was not found.
HISTOPATHOLOGICAL EXAMINATION
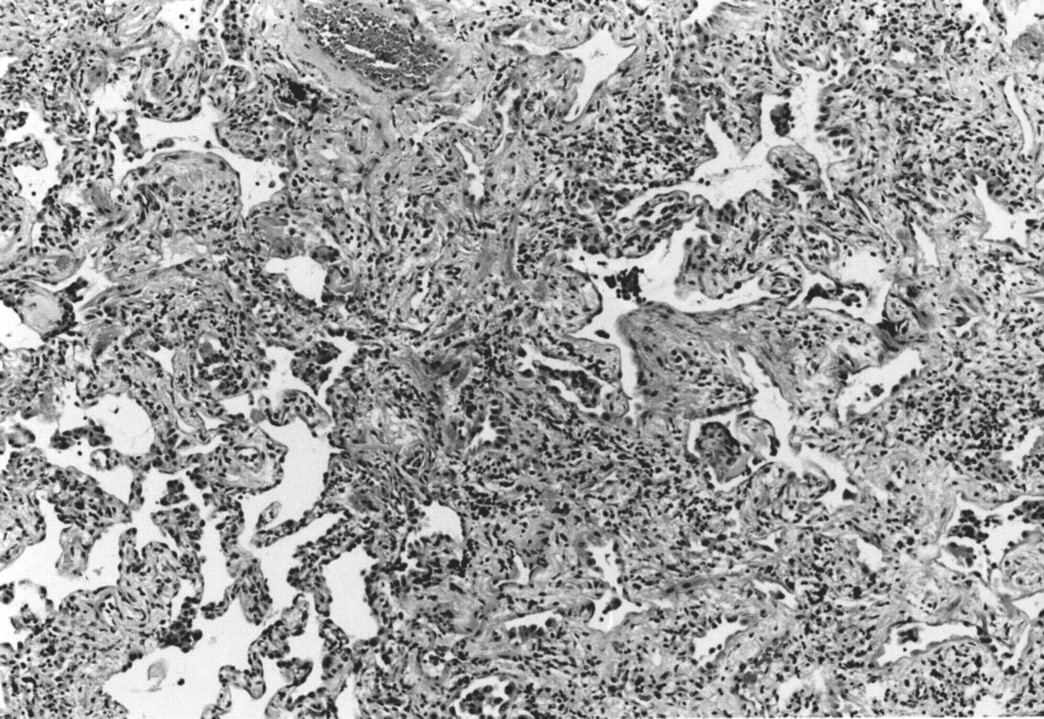
Open lung biopsy specimens of the left lower lobe revealed lung tissue with various degrees of fibrosis. There was no normal lung tissue. The pattern was consistent with usual interstitial pneumonia of chronic idiopathic pulmonary fibrosis (fig 3). Lung tissue cultures were negative for fungi and acid-fast bacilli.
{kind=link}
{kind=link}
{kind=link}
Lung biopsy specimen showing various degrees of fibrosis consistent with usual interstitial type of chronic idiopathic pulmonary fibrosis.
COURSE OF THE DISEASE
When first seen, the patient was started on oral prednisone, 20 mg daily, together with beclomethasone dipropionate and salmeterol inhalers. After the lung biopsy prednisone was tapered down and acitretin, 25 mg/day, was added to the inhalers. After 2 months the patient's breathing had improved and he could walk more and do moderate exercise. The tongue leukoplakia was also improving. There was, however, no improvement in respiratory function tests (FEV1 1.4 l, FVC 1.8 l, FEV1% 77%). In June 1998 the patient was feeling better and his tongue leukoplakia had nearly disappeared, but the respiratory function tests remained unchanged. The platelet count was still in the region of 120 × 109/l. The patient left the UAE to go back to Egypt where he died in February 2000.
His family history disclosed that the patient had two younger brothers, one of whom had some pigmentation on the neck. One maternal uncle had died aged 36 from a gastric carcinoma and another had died also in his mid 30s from a “bleeding problem”. A third brother was alive in his mid 40s but had chest problems, and a fourth, who had died aged 28 in a car accident, had some pigmentation on the neck. Three maternal aunts were alive and healthy and a fourth had had surgery for carcinoma of the cervix. The maternal grandmother, who died aged 72, might have had some pigmentation on the neck. The patient was married and had two healthy children aged 7 and 5 years.
We examined the 35 year old younger brother of the patient. He had a mild reticulate skin pigmentation on both sides of the neck and on the upper chest, with lingual leukoplakia. The left big toenail was dystrophic. Other nails, scalp, facial and body hair were normal. He did not complain of any respiratory problems and chest radiographs were normal. His blood count was normal.
DKC1 MUTATION
The patient and his 35 year old brother presented with missense mutation 5C→T in exon 1 of the DKC1 gene.5 This mutation involves the fifth nucleotide of exon 1, which corresponds to nucleotide 97 from the start of the DKC1 gene. Dyskerin gene mutation 5C→T leads to an alanine to valine substitution of the second amino acid of the dyskerin protein (A2V substitution).
Discussion
The patient presented with several typical mucocutaneous features of DC. The fingernails showed clubbing rather than the expected atrophy, a possible consequence of the longstanding pulmonary disease. There was also moderate thrombocytopenia, and neutrophils and lymphocytes were at the lower end of the normal range. CD8 was raised, CD4 was low, and the CD4/CD8 ratio was inverted. A peripheral cytopenia of at least one lineage has been reported in 93% of 61 patients with DC.6 Isolated thrombocytopenia generally develops in the third or fourth decades of life in survivors. The patient's brother also showed some characteristic features of DC, although to a lesser degree. Although information on the rest of the family was scarce, there may have been more cases of DC among the male relatives.
A salient feature of the patient, however, was his restrictive pulmonary disease with reduced transfer factor and histopathological findings consistent with the diagnosis of usual interstitial pneumonia (UIP) type of idiopathic pulmonary fibrosis.7 UIP has a poor prognosis. Treatment with inhalers improved the patient's breathing comfort but was without effect on respiratory function tests. Retinoids, as expected, improved the lingual leukoplakia.
It has been commented that respiratory disorders reported in association with DC have been poorly documented.8 Our review of the English literature up to 1994 identified nine patients.8-15 Since 1995 patients have been reported in the DC registry established at the Hammersmith Hospital. London6; one additional case was reported in 1998 and is not included in the registry.16 Both patients described here are included in the registry. A tentative classification of pulmonary involvement in DC was made by Imokawa et al in 1994,15 and this has led to the three following questions: (1) What is the incidence of pulmonary disorders in DC? (2) Is there a specific pulmonary manifestation to DC? (3) Is this pulmonary disorder related to specific mutations in the DKC1 gene?
Concerning the incidence of pulmonary disorders in DC, Inoueet al 10 reported a case of DC with pulmonary fibrosis in 1973 and reviewed the literature on 35 cases; they found only one other patient with pulmonary manifestations, also related to fibrotic changes in the lungs.9 In 118 patients from 76 families with affected males only, the DC registry reports pulmonary complications with reduced lung transfer factor and/or restrictive defects in 24, an incidence of 20.3%.1Pulmonary disease in patients with DC may be underestimated. One reason could be that many patients die at an early age. In the registry 67% of deaths resulted directly from bone marrow failure or from complications of its treatment and 80% of deaths occurred before the age of 20.1 Pulmonary disease in DC may occur at an early age but is likely to develop later in life in survivors with absent or mild haematological disease.
Pulmonary disorders such as asthma,14 Pneumocystis cariniipneumonia,12 hepatopulmonary syndrome of Rodriguez-Roisin and Krowka,6 as well as various pulmonary infections (I Dokal, personal communication) have been reported in patients with DC. However, they may present with restrictive pulmonary disease. Whenever available, biopsy or post-mortem histopathological investigations of lung tissue have revealed the common occurrence of interstitial fibrosis. In the patient described here, as well as in two other cases,15 ,16 the interstitial fibrosis was found to be similar to that of UIP. Others have concluded that the risk of restrictive pulmonary disease of unknown aetiology is increased in patients with DC.13 It is interesting to note that three patients in the DC registry died of pulmonary disease following bone marrow transplantation (BMT),6 and it has been assumed that patients with DC are at a high risk of post BMT/graft versus host disease pulmonary complications.1 ,6
It is too early to say whether there is one (or more) specific mutation(s) which predisposes patients with DC to develop pulmonary fibrosis. In the present study we noticed that, although both patients were of a similar age (40 and 35 years), only the 40 year old presented with interstitial pulmonary fibrosis, yet both harboured the same mutation (5C→T) in their DKC1 gene. This mutation has not so far been found in any other patient, and it is also the only mutation reported in exon 1 of the DKC1 gene.5 The DKC1 5C→T mutation does not therefore seem to be responsible for abnormalities leading to pulmonary fibrosis. As advocated for cystic fibrosis,17 it could be that other factors (environmental, genetic or medical) also modulate the course and severity of DC.
It is possible that patients may present with pulmonary disorders as primary complaints in DC and that the condition goes unrecognised because of minor dermatological manifestations, as in the brother of the patient presented here and in the case reported by Bryan and Nixon.18 Genetic diagnosis is now available for these doubtful cases.
Acknowledgments
We wish to thank Drs Inderjeet Dokal and Stuart W Knight for performing genotyping of our patients and for their support of this project.