Article Text

Abstract
Background: Chronic obstructive pulmonary disease (COPD) is a respiratory disorder with increasing prevalence and mortality. It is associated with airway obstruction, increased airway hyper-responsiveness (AHR), and ongoing airway and lung inflammation dominated by CD8 lymphocytes and neutrophils. Single-nucleotide polymorphisms (SNPs) in a disintegrin and metalloprotease 33 (ADAM33) gene have been associated with AHR and COPD.
Objective: To assess whether SNPs in ADAM33 are associated with the severity of AHR and airway inflammation in COPD.
Methods: Eight SNPs in ADAM33 (F+1, Q-1, S_1, S_2, ST+5, T_1, T_2, V_4) were genotyped in 111 patients with COPD (96 males, 69 current smokers, mean (standard deviation (SD)), aged 62 (8) years, median pack-years 42 (IQR 31–55), mean postbronchodilator forced expiratory volume in 1 s (FEV1)% predicted 63 (9). Provocative concentration of methacholine causing a decrease in FEV1 of 20% (PC20 methacholine), sputum and bronchial biopsies were collected.
Results: Patients with the ST+5 AA genotype had more severe AHR, higher numbers of sputum inflammatory cells and CD8 cells in bronchial biopsies than patients with the GG genotype (p = 0.03, 0.05 and 0.01, respectively). CD8 cell numbers were lower in patients carrying the minor allele of SNP T_1 and T_2, and homozygotic minor variants of SNP S_2 compared with the wild type (p = 0.02, 0.01 and 0.02, respectively).
Conclusions: This is the first study revealing that SNPs in a gene that confers susceptibility to COPD in the general population—that is, ADAM33—are associated with AHR and airway inflammation in COPD. These findings constitute an important step forward in linking gene polymorphisms with COPD pathophysiology, thereby possibly contributing to better treatments for this progressive and disabling disease in the future.
- ADAM33, a disintegrin and metalloprotease 33
- AHR, airway hyper-responsiveness
- COPD, chronic obstructive pulmonary disease
- FEV, forced expiratory volume
- PC20, 20% fall in forced expiratory volume in 1 s
- SNP, single nucleotide polymorphism
Statistics from Altmetric.com
- ADAM33, a disintegrin and metalloprotease 33
- AHR, airway hyper-responsiveness
- COPD, chronic obstructive pulmonary disease
- FEV, forced expiratory volume
- PC20, 20% fall in forced expiratory volume in 1 s
- SNP, single nucleotide polymorphism
Chronic obstructive pulmonary disease (COPD) is highly prevalent and the only disease with increasing mortality rates worldwide.1 The disease is characterised by irreversible air flow limitation and associated with an influx of neutrophils, macrophages and CD8 T lymphocytes in the airways.2,3 Most patients with COPD show airway hyper-responsiveness (AHR),4 an exaggerated airway response to non-specific stimuli resulting in airway obstruction. The severity of AHR is positively associated with inflammation in lung tissue5 and numbers of CD8 cells in bronchial biopsies in COPD.6 One study reported an association of AHR with sputum inflammatory cells in COPD,7 whereas another study did not find this association.8
Cigarette smoking is by far the most important risk factor for COPD but there is a wide range in disease severity, irrespective of the number of pack-years of smoking. Furthermore, only a minority of smokers develop the disease, suggesting that besides smoking, an underlying genetic constitution has a role in the development and severity of COPD. A disintegrin and metalloprotease 33 (ADAM33) gene is a gene of putative interest for COPD. It was first identified as a susceptibility gene for asthma and AHR,9,10 and subsequently as a susceptibility gene for COPD in the general population.11 It is furthermore associated with accelerated decline in lung function in the general11 and asthma population.12
ADAM33 belongs to the ADAM family of membrane-anchored metalloproteinases that has a role in a variety of processes such as cell fusion, adhesion and signalling.13,14ADAM33 is expressed in both airways smooth muscle cells and lung fibroblasts. Although the overall substrates and function of ADAM33 are as yet unknown, it has been shown to be able to cleave α2-macroglobulin,15,16 which has a role in pulmonary defence. It is suggested that ADAM33 is involved in tissue remodelling,17 a physiological process intricately related to airway inflammation, hyper-responsiveness and airway obstruction.
The aim of this study was to test the hypothesis that SNPs in ADAM33 are associated with the severity of both AHR and airway inflammation in sputum and bronchial biopsies of patients with COPD.
METHODS AND MATERIALS
The extended version of the methods section is available in the online data repository. In all, 114 patients with COPD participating in the GLUCOLD study were included.18 Patient characteristics have been described in detail previously.18 In brief, all patients had irreversible air flow limitation and chronic respiratory symptoms, and were current or ex-smokers with at least 10 pack-years of smoking. Patients did not use a course of inhaled or oral corticosteroids within 3 months, or maintenance treatment with these drugs within 6 months before randomisation. None of the patients had a history of asthma. The medical ethics committees of the Leiden University Medical Centre (Leiden, The Netherlands) and the Groningen University Medical Centre (Groningen, The Netherlands) approved the study, and all patients gave their written informed consent.
Pulmonary function tests have been described previously.18 Spirometry and reversibility to salbutamol were measured. Methacholine challenge tests were performed with the 2-min tidal breathing method19 and expressed as the provocative concentration of methacholine causing a fall in forced expiratory volume in 1 s (FEV1) of 20% (PC20).
Sputum induction and whole-sample processing were performed according to a validated technique20 as described in detail previously.18
Details on biopsy collection, processing and immunohistology have been published previously.21 In brief, we collected the two best biopsies out of four paraffin-wax-embedded biopsies per patient, and used specific antibodies against T lymphocytes (CD3, CD4 and CD8), macrophages (CD68), neutrophil elastase, mast cell tryptase (AA1) and eosinophils (EG2).
DNA was extracted from peripheral blood. Genotyping was performed as described previously11 using primers and probes from Applied Biosystems TaqMan SNP Genotyping Assays (Nieuwekerk aan de IJssel, Dordrecht, The Netherlands).
Figure 1⇓ shows the eight SNPs in ADAM33 genotyped: F+1 (G/A), Q-1 (C/T), S_1 (Val–Iso), S_2 (G/C), ST+5 (A/G), T_1 (Met–Thr), T_2 (Pro–Ser), V_4 (C/G). We based the selection of the SNPs on previous associations with AHR, excess decline in FEV1, and/or presence of COPD.9,11,12,22,23
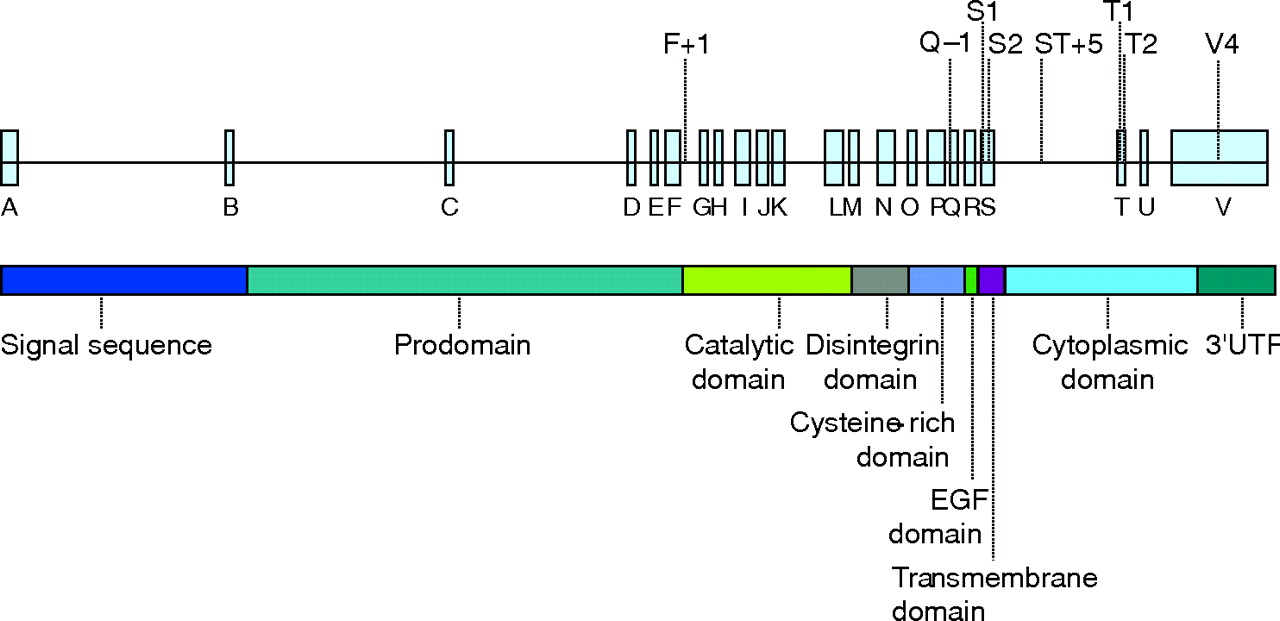
The exon–intron structure of a disintegrin and metalloprotease 33 (ADAM33; the eight genotyped single nucleotide polymorphisms are indicated above the gene) and the domain organisation of ADAM33. EGF, epidermal growth factor; Prodomain, proteinase domain.
We used Arlequin (V.2.000) to test whether SNPs were in Hardy–Weinberg equilibrium and linkage disequilibrium. We investigated whether ADAM33 SNPs are associated with PC20 and the number and subset of inflammatory cells in sputum and bronchial biopsies. Our primary genetic model for all SNPs was a dominant model (homozygotes and heterozygotes for the minor allele being compared as a group with homozygotes for the major allele). In addition, SNPs with a minor allele frequency of ⩾0.30 were entered in (1) a codominant model (three genotype groups per SNP separately) and (2) a recessive model (homozygotes and heterozygotes for the major allele being compared as a group with homozygotes for the minor allele). PC20 and inflammatory cells in sputum and bronchial biopsies were log transformed to obtain a normal distribution. We performed univariate analyses using the t test and ANOVA. Multiple linear regression analyses were performed to investigate the association of polymorphisms in ADAM33 with PC20, sputum inflammatory cells and inflammatory cells in biopsies as dependent variables. Independent variables included in the model were sex, smoking status, lung function, genotype and the interaction of smoking status and genotype. To determine whether the regression estimates (with standard errors) and p values were not due to chance, we performed bootstrapping by resampling rows of the original data frame (using n = 5000 bootstrap replicates) on the main significant outcome variables.
RESULTS
Results of univariate analyses and multiple regression analyses assuming a recessive model are shown in the online data supplement. Multiple regression analyses assuming a dominant and codominant model are presented below. The low number of patients per haplotype did not allow haplotype analysis.
Prevalence of ADAM33 SNPs
DNA was available from 111 out of 114 patients with COPD. Table 1⇓ presents the clinical characteristics. All genotyped SNPs were in Hardy–Weinberg equilibrium and in significant linkage disequilibrium. Table 2⇓ shows the prevalence of the eight SNPs. SNPs ST+5, F+1 and S_2 had a minor allele frequency of ⩾0.30; therefore, these SNPs were analysed both in a dominant model and codominant model. The frequency of SNP ST+5 in our group was 50% for each allele. We use the term AA genotype for the wild type and GG genotype for the homozygotic mutant genotype, as published previously.23
Clinical characteristics
Prevalence of a disintegrin and metalloprotease 33 single nucleotide polymorphisms in patients with chronic obstructive pulmonary disease
Multivariate association of ADAM33 SNPs with AHR
Dominant model: patients with a G-allele for SNP ST+5 had a significantly higher PC20 compared with the AA genotype (geometric mean (GM) 0.61 v 0.27 mg/ml, p = 0.04). SNPs F+1, Q-1, S_1, S_2, T_1, T_2 and V_4 were not significantly associated with the level of AHR.
Codominant model: patients with the GG genotype for SNP ST+5 had a significantly higher PC20 compared with the AA genotype (fig 2A⇓). SNPs F+1 and S_2 were not significantly associated with the level of AHR.

Association of a disintegrin and metalloprotease 33 single nucleotide polymorphism (SNP) ST+5 with airway hyper-responsiveness (provocative concentration of methacholine causing a decrease in forced expiratory volume in 1 s (FEV1) of 20% (PC20 methacholine)), sputum inflammatory cells and CD8 cells in bronchial biopsies in patients with chronic obstructive pulmonary disease. (A) PC20 methacholine per SNP ST+5 genotype. (B) Total sputum cell count per SNP ST+5 genotype*. (C) The number of CD8 cells in bronchial biopsies per SNP ST+5 genotype. *Total sputum cell count refers to the total non-squamous cells in induced sputum. Solid squares represent the geometric mean; vertical bars represent the 95% CI. Different numbers for the SNP genotypes are due to missing genotype data.
Multivariate association of ADAM33 SNPs with inflammatory cells in sputum
Dominant model: patients with one or more G-alleles of SNP ST+5 had a significantly lower total cell count in sputum than the AA genotype (GM 176 v 287×104 cells/ml; p = 0.04). Individuals with minor alleles for SNPs F+1, Q-1 and S_2 had lower numbers of sputum neutrophils compared with the wild type for those SNPs (F+1 GM 103.0 v 134.9 ×104 cells/ml, p = 0.05; Q-1 GM 84.5 v 122.5×104 cells/ml, p = 0.01; S_2 GM 94.0 v 127.9×104 cells/ml, p = 0.02). SNPs S_1, T_1, T_2 and V_4 were not significantly associated with the number of sputum inflammatory cells.
Codominant model: patients with the GG genotype for SNP ST+5 had a significantly lower total cell count in sputum than the AA genotype, and patients with the AG genotype tended to have a lower total cell count (fig 2B⇑).
Heterozygotic individuals for SNP S_2 had higher numbers of sputum macrophages than the wild type (S_2 GG: GM (95% CI) 27.7×104/ml (20.3 to 37.8); GC: 39.5×104/ml (28.7 to 54.5), p = 0.03; CC: 29.6×104/ml (17.6 to 49.7); p = 0.80). Heterozygotic individuals for SNP S_2 also had lower numbers of neutrophils (S_2 GG: GM (95% CI) 127.9×104/ml (97.5 to 168.3); GC: 93.3×104/ml (70.5 to 123.9), p = 0.03; CC: 97.1×104/ml (61.5 to 153.5); p = 0.24).
Multivariate association of ADAM33 SNPs with inflammatory cells in bronchial biopsies
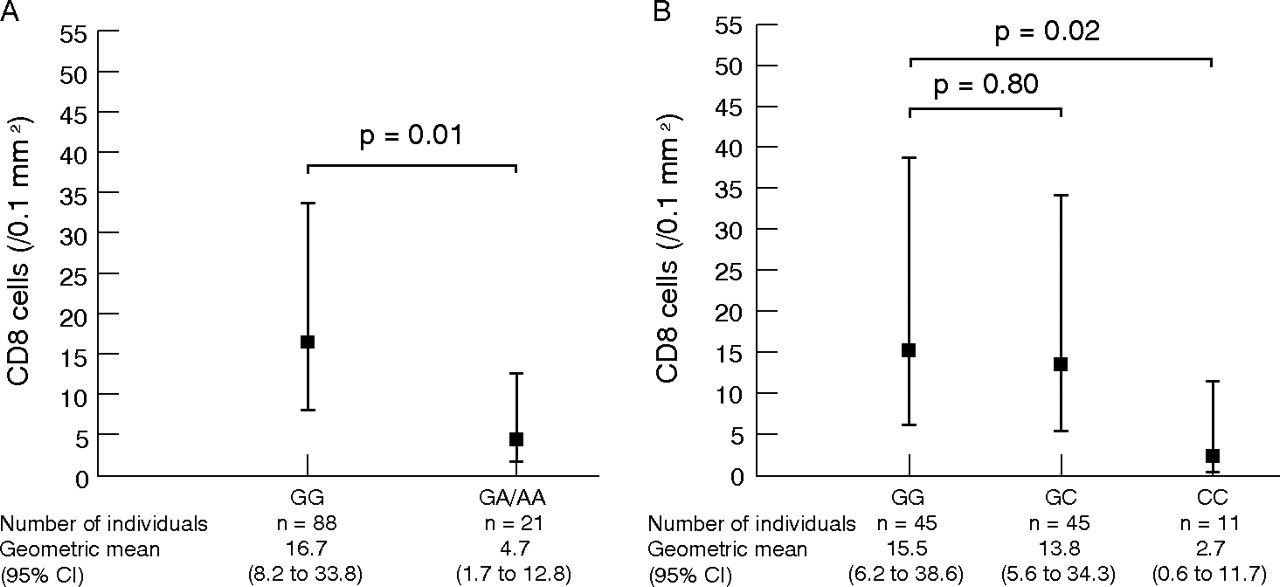
Dominant model: patients with a minor allele for SNPs T_1 and T_2 had a significantly lower number of CD8 cells than the wild type (T_1: p = 0.02; T_2: p = 0.01). We present only the results of SNP T_2 in fig 3A⇓ given the fact that SNPs T_1 and T_2 are in complete linkage disequilibrium (p = 10−16; see online data repository, fig E1) and the association of these SNPs with the number of CD8 cells in bronchial biopsies was similar.
{kind=link}
{kind=link}
{kind=link}
Association of single nucleotide polymorphisms (SNPs) in a disintegrin and metalloprotease 33 with the number of CD8 cells in bronchial biopsies in chronic obstructive pulmonary disease. (A) Per genotype for SNP T_2. (B) Per genotype for SNP S_2. Solid squares represent the geometric mean; vertical bars represent the 95% CI. Different numbers for the SNP genotypes are due to missing genotype data.
Codominant model: patients with the GG genotype for SNP ST+5 had significantly lower numbers of CD8 cells in bronchial biopsies than the AA genotype (fig 2C⇑). Homozygotic individuals for the minor allele of SNP S_2 had significantly lower numbers of CD8 cells in bronchial biopsies compared with the wild type (fig 3B⇑) and lower numbers of plasma cells (S_2 GG: GM (95% CI) 13.5/0.1 mm2 (8.9 to 20.7); GC: 11.8/0.1 mm2 (7.7 to 18.2), p = 0.54; CC: 6.5/0.1 mm2 (3.3 to 12.9); p = 0.04). Heterozygotic patients for SNP S_2 had significantly lower numbers of CD4 cells compared with the wild type (S_2 GG: GM (95% CI) 28.5/0.1 mm2 (19.9 to 41.0); GC: 21.8/0.1 mm2 (16.9 to 28.2), p = 0.04; CC: 25.6/0.1 mm2 (17.0 to 38.3); p = 0.59). No significant associations were found with SNP F+1.
We found no significant associations between SNPs in ADAM33 and the number of mast cells or eosinophils in bronchial biopsies.
Results of bootstrapping confirmed the above-presented main outcome results, with comparable and significant p values.
DISCUSSION
The important message of this study is that SNPs in ADAM33 are associated with the pathophysiology of COPD. Patients with the AA genotype for SNP ST+5 had more severe AHR, higher numbers of sputum inflammatory cells and higher numbers of CD8 cells in bronchial biopsies than those with the GG genotype. Moreover, individuals with the minor allele of SNP T_1 and T_2, and homozygotic individuals for the minor allele of SNP S_2 had significantly lower numbers of CD8 cells in bronchial biopsies, cells relevant to the pathology of COPD.
Prevalences of the ADAM33 SNPs found in our COPD population are comparable to those recently reported by van Diemen et al.11 We also confirm their findings that minor alleles for SNPs F+1, S_1 and S_2 are more prevalent in patients with COPD than in healthy participants. Regarding SNP Q-1 we showed a significantly higher prevalence of the minor allele in patients with COPD compared with healthy controls, whereas van Diemen et al11 showed a trend in the same direction (see online data repository). In addition, we found a higher prevalence for the A-allele in SNP ST+5 in patients with COPD. As far as we know, this is the first study suggesting that a COPD susceptibility gene is also associated with the pathophysiological process in COPD.
ADAM33 is a member of the ADAM family, a group of membrane anchored metalloproteases that contain a disintegrin and a metalloprotease domain and is expressed in multiple tissues, including lung tissue.24 As its function has not been unravelled yet, we can only speculate about its role in COPD. It is conceivable that ADAM33 has a role in both remodelling and inflammation by shedding growth factors, cytokines and their receptors from the cell surface. If a SNP alters ADAM33 function to increase protein production, this might result in enhanced airway inflammation. Alternatively, when ADAM33 has a diminishing effect on the release of proinflammatory cytokines, inflammation would increase by a decrease in ADAM33 production or function. The same could be true for the role of ADAM33 in airway remodelling, in case a change in ADAM33 results in an increase in growth factors resulting in proliferation of airway smooth muscle cells and lung fibroblasts. Whatever the functions of ADAM33 turn out to be, our data suggest that it is associated with inflammation and AHR.
We showed an association between ADAM33 and the severity of AHR in COPD. AHR is important to COPD given its association with accelerated FEV1 decline25 and increased risk of COPD mortality.26 The exact pathophysiology underlying AHR is unclear, but it is thought to result from an inflammatory process in the airways in addition to geometric changes owing to airway remodelling. As discussed above, it is possible that ADAM33 has a role in both these processes thereby contributing to the severity of AHR.
ADAM33 SNP ST+5 is associated with the total sputum cell count in our COPD population. When we replaced this by numbers of sputum neutrophils or macrophages, we found significant associations with SNPs S_2, F+1 and Q-1 but not with the other SNPs. This suggests that not one specific cell type was predominantly accounting for the association of total cell count with SNP ST+5. Several studies have shown an increase in the number of neutrophils and macrophages and concentrations of proinflammatory cytokines like interleukin 8 and tumour necrosis factor α in induced sputum of patients with COPD.27–,29 Intuitively, one would think that the severity of AHR is associated with the degree of inflammation and, in fact, we previously did show an independent, positive association between severity of AHR and total sputum cell counts in these patients with COPD30; perhaps ADAM33 is the missing link.
O’Shaughnessy et al31 showed an increased number of neutrophils and CD8 lymphocytes in bronchial biopsies of smokers with air flow limitation, and this increase was inversely associated with the level of lung function. We, therefore, hypothesised that individuals with a genetic predisposition for a higher number of CD8 cells were more susceptible to a further increase in CD8 cells, which might finally result in air flow limitation. Interestingly, we found an association of SNP ST+5 with both the presence of COPD (see online data repository) and the number of CD8 cells in our patients with COPD.
ADAM33 has previously been associated with asthma in some,9,22,23 but not all studies.32,33 We would like to emphasise that we are confident that our patients with COPD genuinely have COPD and no asthma given the fact they all had moderate to severe air flow limitation after bronchodilation, ⩾10 pack-years of smoking, and no history of doctor-diagnosed asthma. We selected eight SNPs in ADAM33 based on previous literature. One could argue that we did not genotype all known SNPs in ADAM33. However, we did find consistent and significant associations between the genotyped SNPs and the severity of AHR and airway inflammation in our COPD population, indicating linkage of ADAM33 with pathophysiological features of COPD.
A potential criticism of our study is that the sample size of 111 patients with COPD is relatively small. From a practical point of view, biopsy studies are very demanding and the issue of sample size would especially be of concern in case we did not find any associations. However, we did find significant associations between ADAM33 and features of COPD. We set out with the a priori hypothesis that the same SNPs in ADAM33 previously shown to be associated with COPD are also associated with its pathophysiology. To investigate our hypothesis, we performed several analyses to assess the association of ADAM33 with hyper-responsiveness and airway inflammation and found associations of moderate significance. This has affected the interpretation of the results. One could raise the issue of multiple testing being responsible for the current results and that we should have adjusted for this in our analyses. We do not agree with this for several reasons. Firstly, the independent variables in our analyses (eg, sputum total cell count and differential cell count) are mutually related, indicating that a rigid statistical procedure like a Bonferroni correction for multiple testing would not do justice to their biologically linked nature. Secondly, we did not randomly test for associations between ADAM33 and features of COPD but had a predefined hypothesis based on previous literature.
ADAM33 is a highly polymorphic gene containing at least 58 SNPs of which we investigated eight, based on previous literature.9,11,12,22,23 We found an association of SNP ST+5 with the severity of AHR and airway inflammation in induced sputum and bronchial biopsies in COPD. The ST+5 SNP is an intron SNP between the S-exon (transmembrane region) and the T-exon (which includes a SH3 domain and a phosphorylation site). This may have functional relevance, as non-coding introns can exert their effect by influencing alternative splicing, splicing efficiency or messenger RNA turnover.
We furthermore found an association of SNP S_2 with the inflammatory cell profile in sputum and the number of inflammatory cells in bronchial biopsies. A higher number of airway wall CD8 cells is associated with more severe AHR,6 by itself a risk factor for accelerated lung function loss.25 van Diemen et al11 showed an association of the minor allele of SNP S_2 with accelerated lung function decline in a general population and of the minor allele of SNP T_2 with the presence of COPD. Yet, in this study the minor alleles of these SNPs were associated with a lower number of CD8 cells. How can we reconcile these seemingly discordant findings? SNP S_2, a silent mutation, is located in the S-exon and SNP T_2 is located in the T-exon. Both SNPs are in close proximity to SNP ST+5. The significant linkage disequilibrium between SNPs S_2, ST+5 and T_2 may suggest that the part of ADAM33 involved in genetic susceptibility and pathophysiology of COPD lies in the 3′ region. Alternatively, it may still be either at a different location within ADAM33 or at an adjacent gene. Our study was not designed to discuss this question and clearly, it needs further research.
In conclusion, our study confirms ADAM33 as a COPD susceptibility gene and is the first to extend this observation by revealing an association of ADAM33 with the severity of both AHR and airway inflammation in individuals affected with COPD. These findings constitute an important step forward in linking gene polymorphisms with COPD pathophysiology, thereby possibly contributing to better future treatments for this progressive and disabling disease.
Acknowledgments
The GLUCOLD Study Group consists of HF Kauffman, D de Reus, Department of Allergology; HM Boezen, DF Jansen, JM Vonk, Department of Epidemiology; MDW Barentsen, M Luinge, W Timens, M Zeinstra-Smit, Department of Pathology; AJ Luteijn, T van der Molen, G ter Veen, Department of General Practice; MME Gosman, MP Farenhorst, NHT ten Hacken, HAM Kerstjens, MS van Maaren, DS Postma, CA Veltman, A Verbokkem, I Verhage, HK Vink-Klooster, Department of Pulmonology; University Medical Centre Groningen and University of Groningen, Groningen, The Netherlands. JB Snoeck-Stroband, H Thiadens, Department of Public Health and Primary Care; JK Sont, Department of Medical Decision Making; I Bajema, Department of Pathology; J Gast-Strookman, PS Hiemstra, K Janssen, TS Lapperre, KF Rabe, A van Schadewijk, J Smit-Bakker, PJ Sterk, J Stolk, ACJA Tiré, H van der Veen, MME Wijffels and LNA Willems, Department of Pulmonology; Leiden University Medical Centre, Leiden, The Netherlands; T Mauad, University of Sao Paulo, Sao Paulo, Brazil.
REFERENCES
Footnotes
Published Online First 7 November 2006
Funding: This work was supported by Netherlands Organisation for Scientific Research (NWO), The Netherlands Asthma Foundation (NAF; 37.97.74; NAF3.2.02.51), GlaxoSmithKline (NL), Leiden University Medical Centre (LUMC), and University of Groningen (RUG).
Competing interests: None.
Linked Articles
- Airwaves