Article Text

Abstract
Advances in molecular biology have increased our knowledge of the biology of preneoplastic lesions in the human lung. The recently published WHO lung tumour classification defines three separate lesions that are regarded as preinvasive neoplasia. These are (1) squamous dysplasia and carcinoma in situ (SD/CIS), (2) atypical adenomatous hyperplasia (AAH), and (3) diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH). SD/CIS is graded in four stages (mild, moderate, severe, and CIS), based upon the distribution of atypical cells and mitotic figures. Most airways showing SD/CIS demonstrate a range of grades; many epithelia are hard to assess and the reproducibility of this complex system remains to be established. Detailed criteria are, however, welcome and provide an objective framework on which to compare various molecular changes. Alterations in gene expression and chromosome structure known to be associated with malignant transformation can be demonstrated in CIS, less so in dysplasias, but also in morphologically normal epithelium. The changes might be sequential, and their frequency and number increase with atypia. Less is known of the “risk of progression” of SD/CIS to invasive “central” bronchial carcinoma. It may take between one and 10 years for invasion to occur, yet the lesion(s) may be reversible if carcinogen exposure ceases.
AAH may be an important precursor lesion for peripheral “parenchymal” adenocarcinoma of the lung: the “adenoma” in an adenoma–carcinoma sequence. There is good morphological evidence that AAH may progress from low to high grade to bronchioloalveolar carcinoma (BAC; a non-invasive lesion by definition). Invasion then develops within BAC and peripheral lung adenocarcinoma evolves. The molecular events associated with this progression are not well understood and studies are hampered by a lack of clear criteria to distinguish high grade AAH from BAC. Nonetheless, as with SD/CIS, the patterns of expression of tumour associated genes are consistent with neoplastic progression. We have little idea of the incidence of AAH in the normal or “smoking” populations. It is found more frequently in cancer bearing lungs, especially in those with adenocarcinoma, and is more common in women. No data are available on the risk of progression of AAH. DIPNECH is an exceptionally rare lesion associated with the development of multiple carcinoid tumours. Almost nothing is known of its biology.
Knowledge of these lesions will be crucial in the design and understanding of lung cancer screening programmes, where it is likely that the morphological and, more importantly perhaps, the molecular characteristics of these lesions will provide useful targets for detection and possibly even treatment.
- lung cancer
- preneoplasia
- carcinogenesis
Statistics from Altmetric.com
With recent advances in molecular biology, the advent of microdissection techniques, and a greater understanding of the genomic changes involved in malignant transformation, there has been a resurgence of interest in the genesis of primary lung cancer.
Lung cancer is the most common fatal malignancy world wide and global case numbers are set to rise as the smoking epidemic takes hold in Asia, particularly in China. Recent reports confirm that this disease remains very common in the UK and, in Scotland in particular, it is the major cause of death from cancer in both sexes.1 However, the origins of this common disease are not well understood. The progression of disease through squamous metaplasia and carcinoma in situ in the bronchi has been long recognised,2, 3 but it is only recently that some of the molecular and genetic changes associated with the morphological transformation have been elucidated.
But how many carcinomas of the lung arise in this way? It seems likely that the vast majority of squamous cell cancers and probably most others that arise in the central airways do so. However, peripheral lung cancers, predominantly adenocarcinoma, might arise through a different mechanism and a different series of progenitor or preinvasive lesions.
The concept of “scar cancer” is long established, based upon observations that adult lungs often show peripheral scars (old tuberculosis, infarcts, and other inflammatory foci) and peripheral lung tumours frequently possess central scarring, which is believed, by some, to have induced the tumour. More recent studies have suggested that this is not the way in which most peripheral cancers arise. On the basis of morphological and radiological observations, survival data, and studies of collagen types in scars,4–6 the prevailing view is that, in most peripheral tumours (largely adenocarcinoma) with central scarring, the fibrosis postdates the development of thetumour. Shimosato presented a critique of the scar cancer hypothesis containing the following points4:
-
Most adenocarcinomas arise in areas of lung that show no evidence of fibrosis.
-
The central fibrotic scar is usually smaller than the tumour and might also be present in metastases from the same tumour, including lymph node deposits.5
-
Metastatic lung tumours can have central scars.
-
Psammoma bodies present in the central scar point to the earlier presence of tumour.
-
Radiological evidence of cicatrisation is usually seen after the tumour becomes visible on the chest radiograph.
-
The relatively poor prognosis of tumours with central scars might imply that the lesion has a longer natural history.
Nonetheless, several diffuse fibrosing conditions are undoubtedly associated with an increased incidence of lung cancer and probably an excess of adenocarcinoma. Although this association with diffuse fibrosis probably accounts for only a small number of cases overall, various studies on postmortem and surgically resected material found lung cancer in between 10% and 20% of lungs showing diffuse pulmonary fibrosis or honeycomb lung.7 The association is made between tumour development and the hyperplasia of alveolar lining cells, which may be atypical, seen in these conditions. Why patients with systemic sclerosis should be particularly at risk is not clear. Other causes of diffuse lung fibrosis include asbestos and silica and these factors are also linked to the development of lung cancer. It seems, however, that with asbestos, fibrogenesis and carcinogenesis might have independent mechanisms.8 The same may be true for silica exposure. Fibrosis associated with silica exposure (silicosis) appears to be associated with an excess risk of lung cancer but, for exposure to silica without fibrosis, the evidence is tenuous.9 The topic of scar cancer has been reviewed elsewhere.10
More recently, considerable attention has been paid to atypical adenomatous hyperplasia (AAH) as a preinvasive lesion, as the adenoma in a putative “adenoma–carcinoma” sequence in the lung periphery,11, 12 leading to the development of bronchioloalveolar carcinoma and invasive peripheral adenocarcinoma of the lung. Even less is understood regarding the origins of pulmonary neuroendocrine tumours, although there is some evidence that some carcinoid tumours might be related to focal bronchial neuroendocrine cell hyperplasia.
In this review, I will describe the recognised pulmonary preneoplastic lesions, bronchial squamous dysplasia/carcinoma in situ (CIS), AAH, and neuroendocrine cell hyperplasia, and their morphology. I will then consider the progression of squamous dysplasia/CIS to invasive disease, including what is understood of the molecular basis of this progression. Finally, I will discuss what is known of the probable progression of AAH to adenocarcinoma. Nothing is known about the molecular events that might occur in neuroendocrine cell hyperplasia.
WHO classification of preinvasive lesions in the lung
The recently published third edition of the World Health Organisation (WHO) lung tumour classification13 lists three main forms of preinvasive lesion in the lung: (1) squamous dysplasia and CIS, (2) AAH, and (3) diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH). The morphology of these are described in turn.
SQUAMOUS DYSPLASIA/CIS
Many reports refer to the grading of dysplasia of squamous epithelium in the bronchi as being similar to that in other sites such as the cervix or oesophagus. Some authors refer to the usual mild, moderate, and severe atypias, as well as CIS, whereas others tend to lump the grades together, or refer instead to low grade and high grade preinvasive disease, the latter category including CIS. The WHO classification provides criteria for the distinction of mild, moderate, and severe dysplasia and CIS. These are summarised below and presented in an abbreviated diagrammatic form in fig 1.

A diagrammatic summary of the WHO criteria13 for bronchial squamous dysplasia/carcinoma in situ (CIS). The distribution of various features is related to the lower, middle, and upper thirds of the multilayered “squamous” epithelium.
Mild dysplasia
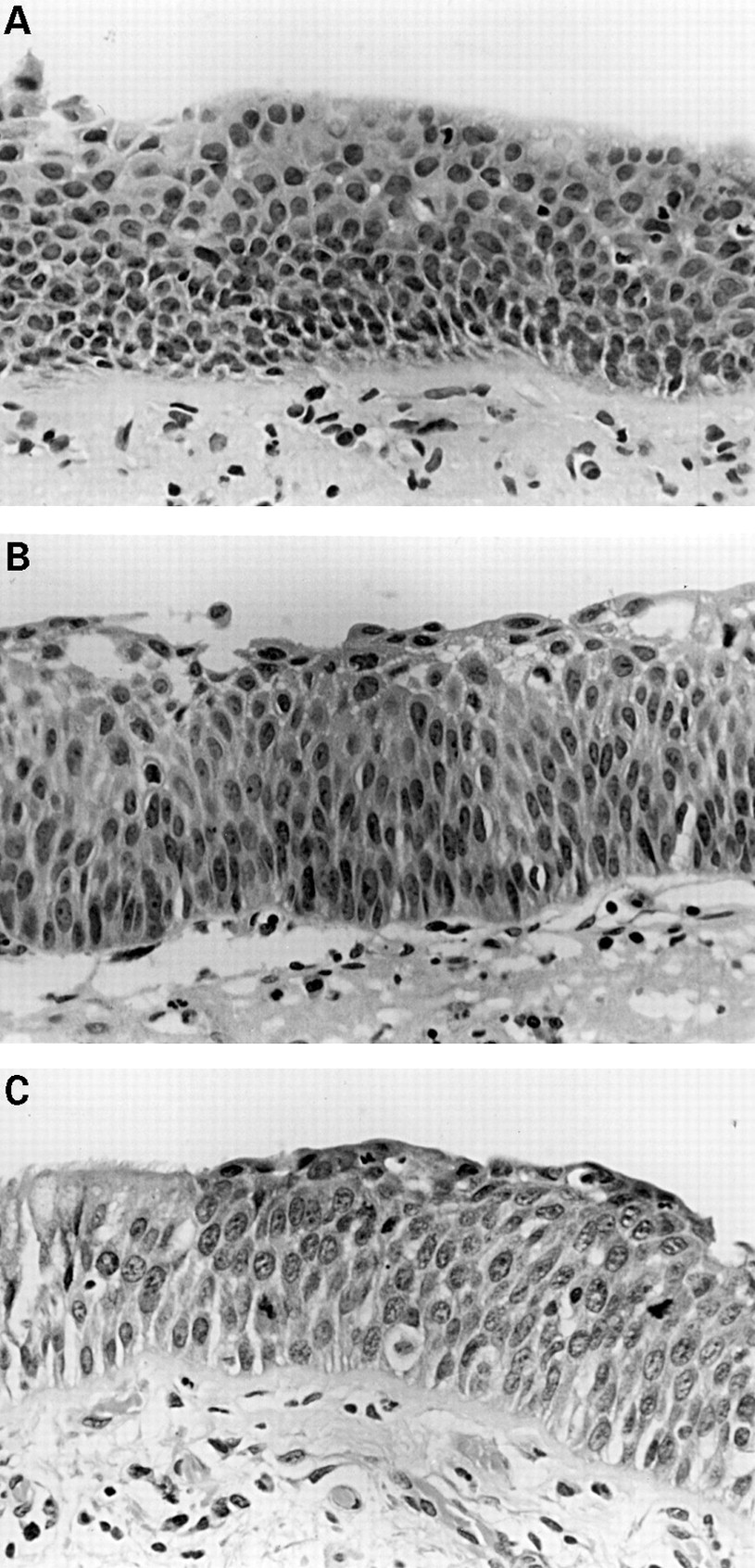
Mild dysplasia is present when the architectural and cytological disturbance is minimal (fig 2A). Basilar zone expansion with cellular crowding is limited to the lower third of the epithelium, as are vertically oriented nuclei. Mitoses are absent. This would occur in an epithelium that is clearly squamous and is distinct, in this classification, from basal cell hyperplasia. The latter is present when the basal epithelial cell layer is more than three cells thick and may well be a precursor of (immature) squamous metaplasia.

(A) Mild dysplasia: basal zone crowding and minimal cytological atypia. (B) Moderate dysplasia: vertically orientated nuclei occupy the lower two thirds of the epithelium whereas maturation is still clearly visible towards the surface. (C) Severe dysplasia: greater cytological aberration, mitoses are clearly present in the lower two thirds of the epithelium, and there is virtually no maturation. Note the sharp transition from normal respiratory epithelium on the left.
Moderate dysplasia
Moderate dysplasia exhibits more cytological irregularity but still has finely granular chromatin. Maturation of cells from the base to the luminal surface shows only partial progression, but there is still clear flattening of the superficial cells (fig 2B). The basilar zone now occupies two thirds of the epithelium, as do vertically oriented nuclei, and mitoses are present in the lower third.
Severe dysplasia
Severe dysplasia exhibits considerable cellular pleomorphism, coarse, uneven chromatin, and little cell maturation, but still shows superficial cell flattening. Basilar zone crowding extends well into the upper third, vertical nuclei and mitotic figures are present in the lower two thirds (fig 2C), and the prickle cell layer has almost disappeared.
Carcinoma in situ
CIS may or may not be associated with epithelial thickening. Cytological aberration is extreme, mitoses occur at all levels, and maturation is absent such that, if the epithelium were inverted, it would not look different (fig 3). Although this is the usual form of CIS, with a flat topography, a more unusual form exists where the in situ lesion develops into an exophytic, polypoid, or papillary growth that can cause mechanical or functional airway obstruction, despite the absence of mucosal invasion.

Carcinoma in situ. Full thickness severe cytological atypia with a chaotic appearance.
There can be considerable overlap between these four categories and in any particular case a range of grades may be seen. These lesions are infrequently encountered by the surgical histopathologist in endoscopic biopsy material. They do, however, shed cytologically atypical cells into sputum and bronchial brushings and washings, although here their importance can be difficult to determine. Bronchial squamous dysplasia/CIS is relatively frequent in lung resection specimens from cigarette smokers with lung cancer but, in this context, reporting and classification of the lesions is less important, outside of any academic considerations. Given that, on conventional bronchoscopy, most of these preinvasive lesions are not visible, their appearance in bronchial biopsy specimens is fortuitous. If fluorescence bronchoscopy techniques, such as LIFE (lung imaging fluorescence endoscopy), become more widely used, the situation might change.14, 15 Such techniques greatly increase the sensitivity of bronchoscopy in finding areas of abnormal epithelium, based on their tendency to show less autofluorescence than normal mucosa. Unfortunately, the specificity of this technique is not high and perhaps only one third of all biopsies taken from areas signalled as “abnormal” during LIFE show any histological abnormality. However, histological normality does not guarantee the absence of genomic alterations in bronchial epithelial cells (see below).
Until these WHO criteria are widely read, tried, and subjected to tests of reproducibility, we will not know how useful this classification is. The authors acknowledge that lack of experience and ill defined boundaries between categories will lead to “problems with reproducibility” and that “the clinical significance of the grading system remains to be established”. It is perhaps surprising, especially if you are a “lumper” rather than a “splitter”, that four separate categories, including a distinction between severe dysplasia and CIS, were defined, rather than two (low versus high grade dysplasia), which seems to be the current trend. Nonetheless, we now have criteria for these four grades. Time will tell us whether or not this was the correct choice.
Another factor also needs to be taken into account. This process is one that does not necessarily affect the full thickness of the epithelium. The process of squamous metaplasia/dysplasia appears to start in a zone of pre-existing basal cell hyperplasia. Squamous differentiation may occur in this basal cell zone still surmounted by differentiated respiratory glandular epithelium; so called immature squamous metaplasia. This can, in turn, develop atypia, and it is as common to find areas of quite pronounced atypia beneath layers of ciliated and mucus secreting columnar cells as full thickness squamous change itself (fig 4A and B). The criteria for assessing dysplasia assume a full thickness squamous epithelium. Grading dysplasia in this partially transformed epithelium is difficult. My current practice is to ignore the overlying respiratory epithelium and attempt to grade the squamous layer: realistically only a “low” or “high” grade can be given.

(A) Ciliated respiratory epithelial cells overlying a population showing clear squamous differentiation (intercellular bridges) and prominent vertical orientation of nuclei (moderate dysplasia?) (B) Apart from the persisting superficial ciliated cells, this could be regarded as carcinoma in situ.
ATYPICAL ADENOMATOUS HYPERPLASIA
The first descriptions of this lesion and its association with lung adenocarcinoma were made by Shimosato and colleagues in 1982,16 and Japanese workers17 together with the late Roberta Miller in Vancouver laid the foundations for an adenoma–carcinoma sequence in the lung periphery, where the adenoma is the lesion known as AAH.11, 12
These lesions are usually invisible to the naked eye, but the larger ones or those with more fibrosis in their walls may be visible on the cut surface of well inflated and formalin fixed lung (fig 5). Most lesions, however, are incidental microscopic findings. A group of alveoli, not infrequently distributed around a terminal or respiratory bronchiole, is lined by plump cuboidal or low columnar cells with the morphology of type 2 pneumocytes or Clara cells (fig 6). There is, by definition, no generalised fibrosis or inflammation in the area, although the alveolar walls lined by this cell population might be slightly thickened owing to excess collagen and/or lymphocytes. Unusual examples show large amounts of scar tissue or a heavy lymphoid infiltrate. Most lesions (75%) range in size from less than 1 mm to about 3 mm. Larger lesions are unusual, although I have seen examples of what I regard as AAH up to 19 mm. Usually, although not always, the larger lesions show increasing cytological atypia, the cells become columnar and may lose their “hobnail” Clara cell features. Nonetheless, mitotic figures are distinctly unusual. Some cells show nuclear inclusions. From a discontinuous layer of cuboidal cells lining alveolar walls, characteristic of what some call low grade AAH, the more atypical lesions are more cellular, showing a more continuous lining of larger, columnar cells with more cell–cell membrane apposition. Papillary structures may develop and protrude into the alveolar space (fig 6). Macroscopically, the larger lesions may appear cystic because of the loss of the central alveolar structure.

(A) A 4 mm diameter focus of atypical adenomatous hyperplasia (AAH) easily visible on the lung cut surface as a result of abundant collagen in the alveolar walls. (B) AAH lesions are often found in the centriacinar zone, near respiratory bronchioles.

Atypical adenomatous hyperplasia. Low grade lesions show (A) intermittent or (B) complete runs of cuboidal cells lining slightly thickened alveolar walls. More cellular lesions (C) may be larger and have columnar Clara-like cells. Less often, lesions are very cellular, more atypical (D), and very difficult to distinguish from bronchioloalveolar cell carcinoma.
There is an imperceptible point at which a larger and more atypical (high grade) focus of AAH becomes a small bronchioloalveolar cell carcinoma (BAC) (a non-invasive lesion under current WHO classification).13 Distinguishing between the two is almost impossible. Miller arbitrarily suggested that any lesions over 5 mm in diameter should be considered a carcinoma.12 However, this might be too simplistic. Some very large lesions are extremely bland with a discontinuous cell population lining the alveolar walls. Much smaller lesions may show much more cell crowding and atypia. It remains to be seen whether reliable criteria can be produced to distinguish AAH with “high grade” atypia from BAC or whether, in fact, these lesions are one and the same thing.
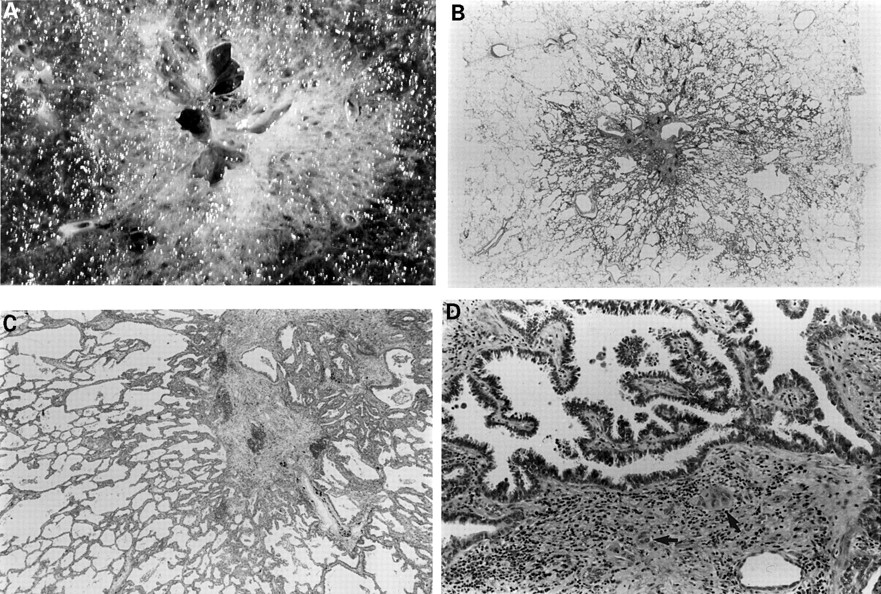
Most AAH lesions have been found incidentally in lung resected for other reasons, primarily lung cancer (see below). Thus, the outcome for the patient has been determined by the fully developed cancer, making it difficult to know how their AAH might progress. The site, size, and invisibility of AAH by most imaging methods makes longitudinal studies of AAH even more difficult than for bronchial dysplasia/CIS. Some AAH lesions seem to become extremely scarred, retaining the architecture of the lesion, but losing part or all of the characteristic cell population lining the alveoli. Other rare examples of what are ostensibly high grade AAH lesions show clear evidence of stromal invasion (fig 7). These lesions are, by definition, adenocarcinoma. Whether this makes the remainder of the lesion BAC or AAH is open to debate. From a morphological perspective there seems little doubt that AAH might progress and develop, through a stage of BAC, into invasive adenocarcinoma.

(A) This 1.7 cm lesion with a rather solid and cystic centre has alveolar spaces clearly visible in the periphery. (B) Whole mount of the same lesion. Closer examination (C) shows a periphery typical of atypical adenomatous hyperplasia but the central, solid zone shows (D) papillary bronchioloalveolar-like areas and clear evidence of stromal invasion (arrows).
NEUROENDOCRINE CELL PROLIFERATIONS AND DIFFUSE IDIOPATHIC PULMONARY NEUROENDOCRINE CELL HYPERPLASIA
Hyperplasia of airway neuroendocrine cells, without invasion beyond the epithelial basement membrane, may be seen in association with chronic inflammation or fibrosis in the lung. When there is extension of these cells interstitially, associated with some fibrous tissue, the lesion is characterised as a carcinoid tumourlet. If such a lesion exceeds 5 mm in diameter, it should be regarded as a typical carcinoid tumour. Such a proliferation of neuroendocrine cells, in association with chronic inflammation, is not regarded as a preinvasive lesion.
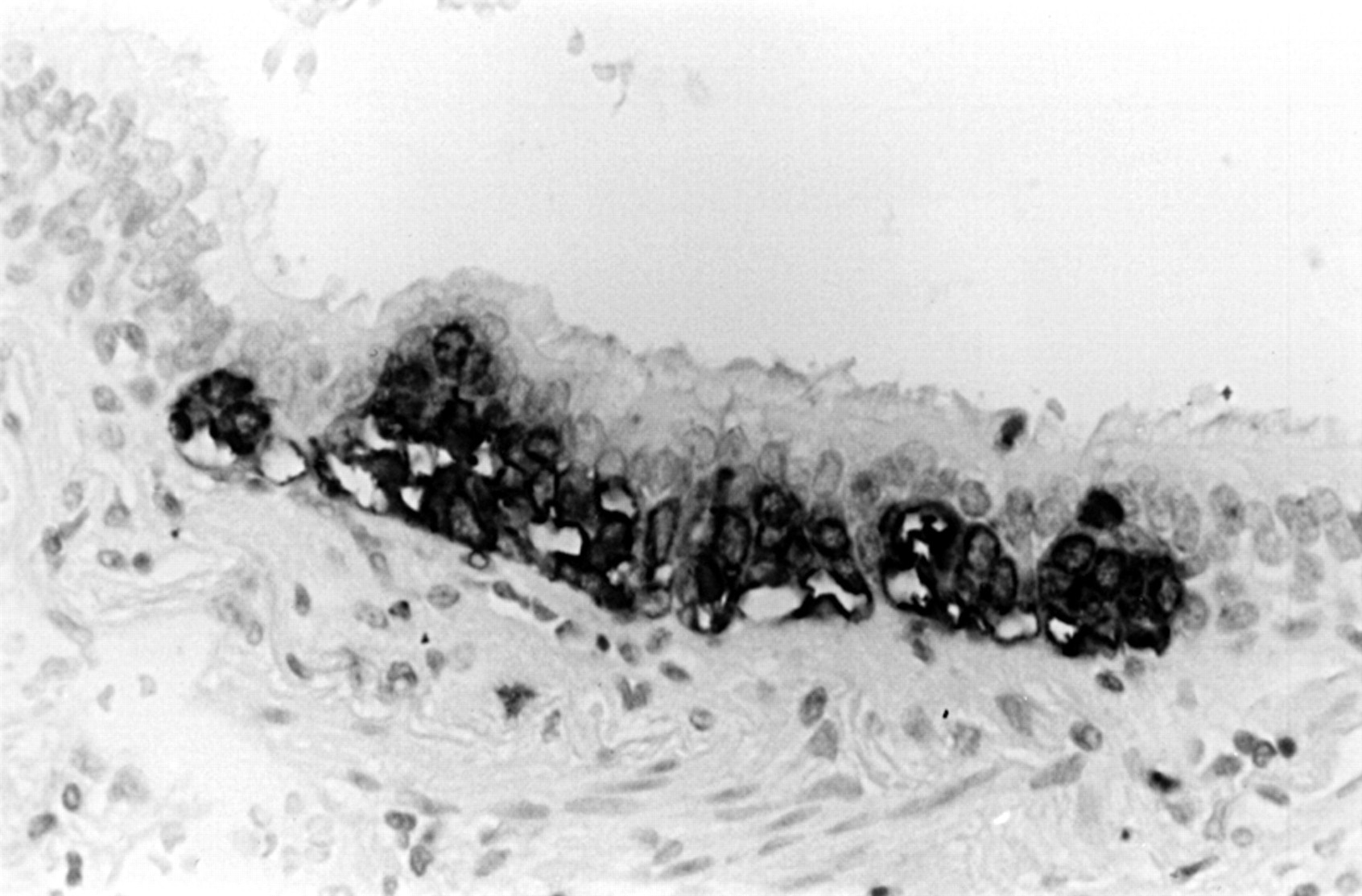
Hyperplasia of neuroendocrine cells is also seen in the airway mucosa in lungs bearing typical (usually peripheral) carcinoid tumours.18 This process may also occur in lungs showing higher grade neuroendocrine malignancy (fig 8). The relation between the hyperplasia and the tumour, if any, is not currently understood.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A hyperplastic focus of neuroendocrine cells in the airway epithelium, found in the same lobe as an atypical carcinoid tumour (immunoperoxidase with anti-chromogranin).
In 1992 Aguayo et al described six unusual patients who had diffuse hyperplasia of airway neuroendocrine cells and evidence of airways obstruction.19 These patients also had multiple pulmonary carcinoid tumourlets. In these and subsequent cases the airway fibrosis, perhaps because of the effects of some paracrine secretion, was presumed to be the cause of the widespread small airways obstruction. As well as multiple carcinoid tumourlets, some of these patients also had one or more carcinoid tumours. Such cases are rare, so experience of this lesion is limited. In the above context, however, it is not unreasonable to suggest that the widespread neuroendocrine cell hyperplasia (DIPNECH) might be a preinvasive lesion, which may give rise to typical carcinoid tumours. What relation this lesion or a similar more localised process might have to most invasive neuroendocrine tumours—from carcinoids (typical and atypical) to the high grade large cell and small cell neuroendocrine carcinomas—is not known. However, it is tempting to speculate that such focal hyperplasia might provide a basis upon which neoplastic transformation is more likely to occur (see below).
SQUAMOUS DYSPLASIA/CIS AND ITS PROGRESSION
Little is known about the rate and risks of progression of squamous dysplasia to CIS and ultimately invasive disease.
There is a large amount of animal experimental work20–22 using a variety of carcinogens and subjects, including the infamous smoking beagles. These data suggest that the earliest change in the epithelium, in keeping with similar observations in epithelia at other sites, is hyperplasia in the basal layer of cells. Such a hyperplasia in the stem cell compartment of the epithelium is one of the earliest steps required in currently accepted theories of multistep carcinogenesis.23 As a result of chronic irritation/stimulation, this expanded cell compartment may differentiate towards a phenotype better adapted to the prevailing environment (cigarette smoke—for example) and squamous metaplasia occurs. Thus, the earliest changes represent a relatively acute response to injury. Over time, a more subacute preneoplastic form of nuclear “injury” takes place which, in turn, leads to the expression of the evolving abnormal genotype as an abnormal phenotype, namely dysplasia and carcinoma in situ.
Experimental studies suggest that all of these changes, up to and including CIS, might be reversible if the stimulus/carcinogen is removed.21, 22 At some point, however, the process (and lesion) become irreversible and we have, by definition, a neoplasm.24 There is also evidence that preinvasive dysplasias may “wax and wane” depending on the dose/duration of exposure to particular carcinogens. Removal of the carcinogens might cause the visible lesion to regress (phenotypic regression); however, the genomic damage (see later) might not disappear and may persist in microscopically apparently normal respiratory epithelium. In more prolonged experiments there is also evidence that the dose and type of carcinogen might determine the type of cancer that develops.21, 22 This is an interesting parallel with the recently observed increase in small cell carcinoma and adenocarcinoma and the fall in squamous lung cancers in the Western world that might, at least in part, be related to changes in smoking habits or in the tobacco or additives used in cigarettes.
Clinical studies of bronchial preinvasive lesions are limited and often involve a population who are “at risk” or the subject of a lung cancer screening programme. Bronchial hyperplasia and squamous metaplasia are common in the airways of cigarette smokers,25 whereas dysplasias and carcinoma in situ are particularly found in cancer bearing lungs. Auerbach et al found dysplasia in 40% of heavy smokers.2, 3 In another study, one third of patients who had atypical cells in sputum went on to develop lung carcinoma.26 In a study of uranium miners, who were subjected to repeated sputum examination, increasingly severe cytological abnormality could be observed in squamous cells with, in some, the development of invasive carcinoma over a period of one to 10 years.27 Up to 20% of worker cohorts exposed to chromates or mustard gas have also been shown to develop preinvasive bronchial changes and to have an increased risk of developing invasive cancer.28
In studies of populations screened using sputum cytology, some individuals will show atypical (squamous) cells. Most of these patients, not including an at risk occupational group, will be cigarette smokers. In such a situation the presence of atypical cells in the sputum usually leads to bronchoscopy, and some studies have shown that it may take six to 36 months for a bronchoscopically visible (standard technique) lesion to appear, if one is not present at the initial examination.29–32
In those patients where atypical sputum cytology leads to bronchoscopy and a lesion is seen (if not at the initial investigation, then at subsequent examination and perhaps after selective bronchial brushings are made in an attempt to localise the source of the abnormal cells to a particular airway), biopsy of the visible lesions yields a range of histological abnormalities. All of these patients would have a normal chest radiograph so that, should malignant cells be detected, these lesions are classified as so called radiographically occult squamous carcinoma of the lung.29–32 In a review of several reported series of such “occult” carcinomas, Carter and Patchefsky found that 25% of the patients had CIS, 42% had early invasive disease confined to the mucosa, whereas 33% showed a small, radiographically invisible cancer that extended beyond the bronchial cartilage.28
It is generally accepted that a tumour has to be at least 1 cm in diameter and unobscured by rib, vascular, or mediastinal shadows to be reliably detectable on a plain chest radiograph. At 1 cm diameter, a tumour has already undergone around 30 doublings of its cell population, something that possibly took several years. Thus, a radiologically visible lung tumour is usually already relatively late in its natural history, having been invasive for months or years before detection, and may already have metastasised. Hence, the failure of most radiograph based screening programmes to improve the cure rate for treating lung cancer. If fluorescence bronchoscopy becomes widely used it is possible that many more early lesions will be found. Even if patients with occult lung cancer are treated by surgery or, more recently, by photodynamic treatment using laser bronchoscopy, the apparent “cure” rates of 80–95% are marred by the finding that up to 25% of patients develop a second lung cancer, sometimes again occult.
Nagamoto et al have made an extensive study of a series of occult lung cancers, including CIS, found through the Miyagi programme of mass screening between 1982 and 1991.30–32 They distinguished between two patterns of occult carcinoma. Most cases (86%) were classified as the “creeping type” where the dominant growth pattern was a horizontal spread within the mucosa, sometimes over several centimetres. The remainder seemed to have a predominantly transmural rather than longitudinal growth pattern (penetrating type). These latter lesions were thought to be more rapidly growing and more likely to become advanced in a short time. However, even the creeping type of lesion showing minimal invasion did have a chance of metastasis,31 although only when the extent of the lesion was longitudinally > 20 mm, when 24% showed nodal spread. No lesion < 20 mm in extent was found to spread. In a study of 19 cases of isolated CIS discovered during the same screening study,32 and treated by surgical resection, no patient had lymph node metastases. Follow up of these patients showed an excellent prognosis. Six of the patients showed a polypoid or micronodular protuberance in the mucosa on bronchoscopy. Such an exophytic and exuberant thickening of the in situ lesion is unusual, but was also described by Spencer et al.33 These lesions may have bronchoscopic, cytological, and even biopsy appearances that are highly suggestive of malignancy. I have seen at least two such cases where, after lobectomy, the preoperative diagnosis of squamous carcinoma required revision to in situ disease. In neither was there evidence of nodal metastatic disease, yet in one case in particular, there was pronounced nodal enlargement, presumably secondary to the considerable “obstructive” pneumonia in the segments distal to the airway containing the polypoid CIS. Even if such lesions are too small to cause mechanical obstruction, functional obstruction might result from the loss of mucociliary function if there is extensive replacement of normal respiratory mucosa by (atypical) squamous epithelium.
MOLECULAR BIOLOGICAL AND GENETIC STUDIES ON SQUAMOUS DYSPLASIA/CIS
Extensive work has been carried out on a range of preinvasive bronchial lesions. This has generally involved either immunohistochemistry, or microdissection of lesional tissue followed by DNA extraction, PCR amplification, and experiments to seek gene mutation or loss of heterozygosity (LOH) of various chromosomal regions.
Cell proliferation and related markers
As might be expected from morphological studies, hyperproliferation, as demonstrated by the use of anti-proliferative cell nuclear antigen (PCNA) antibodies, seems to be an early event in the transforming bronchial epithelium. The proliferative component was shown to rise from around 25% in apparently normal epithelium, through 35–40% in low and high grade dysplasias, to 85–90% in invasive squamous carcinomas, both early and advanced.34 The distribution of positively staining nuclei mirrored the change in epithelial cytoarchitecture from the base to the more superficial layers as atypia increased.35
In a study of Rb, p16, and cyclin D1 expression in bronchial preinvasive lesions, Brambilla et al found that although Rb loss was not seen in dysplasia/CIS, loss of p16 expression was found in moderate dysplasia (12%) and CIS (30%), exclusively in lesions found in cancer bearing lungs, and not in lesions from patients who did not have cancer.36 The same authors found cyclin D1 overexpression in earlier lesions: 6% of hyperplasia/metaplasia, 17% of moderate dysplasia, 46% of moderate dysplasia, and 38% of CIS lesions. It was also overexpressed in preinvasive lesions in 14% of those who did not have cancer.
DNA ploidy
Studies of the DNA content of cell nuclei, as a rough measure of chromosomal gain or loss, have been used to measure the degree of “nuclear aberration” in a malignant cell population. Hirano et al demonstrated aneuploidy in all of the invasive squamous carcinomas tested but not in normal bronchial mucosa.34 Of the preinvasive lesions, 8% of the low grade cases and 33% of the high grade cases were aneuploid. These authors suggest that the development of aneuploidy is a relatively early event in the progression to malignancy and also showed that the development of aneuploidy depended on the pre-existence of a hyperproliferative state in the bronchial epithelium. These graded changes in the nuclei of preneoplastic lesions have also been shown by others, both in retrospective analysis of biopsy material from human patients and in dogs experimentally exposed to various carcinogens.21
bcl-2 expression
The bcl-2 protein is thought to inhibit programmed cell death. Therefore, (over)expression in tumours might give a cell population some survival advantage. Bcl-2 protein has been shown to be overexpressed in lung cancer (25% of squamous carcinomas, 10% of adenocarcinomas, 64% of small cell carcinomas)37–39 and in preinvasive bronchial lesions.40 Katabami et al showed that 33% of bronchial dysplasias expressed Bcl-2 protein, with a weak association with an increasing degree of dysplasia.41 Brambilla et al found bcl-2 overexpression in association with bax downregulation, but this did not correlate with p53 status.42
p53 expression
Various studies have found an increase in stainable p53 protein with increasingly severe bronchial squamous dysplasia (table 1). Several studies describe localisation of p53 protein in nuclei in the zone of the epithelium, which showed loss of nuclear polarity.43, 44 In a large series, Bennett et al also found p53 protein in 6.7% of squamous metaplasias but none in normal epithelium.44 Katabami et al found that in dysplasia in patients with pneumoconiosis, low grade dysplasia (mild/moderate) had a greater tendency to p53 positivity (40%) than in those without pneumoconiosis.41 Brambilla and co-workers found p53 expression in all cases of squamous dysplasia/CIS from cancer bearing lungs, but similar lesions from lungs with no cancer showed no staining, implying that the stabilisation of p53 in the preinvasive lesion had a high predictive value for invasion.42
Stainable p53 protein in bronchial preinvasive disease
The demonstration of stainable p53 protein might infer gene alteration, but the detection of gene mutation might be more important. In a study that included 13 examples of bronchial squamous metaplasia and 22 squamous dysplasias (ungraded), only two of the dysplasias showed evidence of p53 gene mutation.45 Earlier, smaller studies also found evidence of p53 gene mutation, sometimes different from any concurrent tumour, sometimes the same mutation as found in adjacent tumour.46–48
The general conclusion is that even in the very early stages of malignant transformation, in the least dysplastic of the preinvasive lesions, abnormal expression of p53 occurs. This suggests that p53 mutation might be a relatively early event in central bronchial carcinogenesis.
Telomerase
Telomerase is activated and expressed in most human cancers and it may be one mechanism—by preventing progressive telomere shortening, and therefore cellular senescence—that confers immortality to tumour cell populations. Most adult somatic cells have inactive telomerase, but at some stage in the carcinogenic process it becomes reactivated. Yashima et al,49 using in situ hybridisation, demonstrated much greater telomerase positivity in both hyperplastic and dysplastic bronchial epithelium (70–80%) when compared with normal controls (20%), and CIS and invasive disease were positive in 95–100% of cases. However, preinvasive lesions, including CIS, had enzyme activity that was only three to fourfold higher than normal, whereas in invasive disease, telomerase activity was 40 times greater. Once more, even at the earliest stages, telomerase dysregulation appears to occur as part of the multistage pathogenesis of lung cancer.
K-ras mutation
K-ras is more commonly found mutated in adenocarcinoma than in squamous carcinoma. Nonetheless, mutation of K-ras might be of importance in the development of at least some central “bronchogenic” carcinomas arising from the hyperplasia–dysplasia–CIS sequence in the bronchial/bronchiolar epithelium. In a study concentrating primarily on squamous dysplasias in six patients whose tumours (five adenocarcinomas, one squamous carcinoma) all showed a K-ras mutation, Sugio et al were unable to demonstrate K-ras mutation in any normal or hyperplastic epithelium and found only one mutated preinvasive lesion out of the 12 studied.50 This suggests that K-ras mutation might either be a very late or relatively unimportant step in central bronchial carcinogenesis. This is also the conclusion of another study, which found no evidence of K-ras mutation in bronchial adenocarcinoma, a tumour that might arise from bronchial epithelial dysplasia, whereas 35% of peripheral adenocarcinomas, which arise by a different mechanism (see below), showed K-ras mutation.51
Human papillomavirus (HPV)
HPV-6 DNA was found in one of 10 squamous metaplastic lesions by Béjui-Thivolet et al,52 whereas 18% of squamous carcinomas contained HPV DNA from several subtypes, predominantly HPV-18. No cases of preinvasive disease with dysplasia were studied and other work suggests that HPV has no important role to play in the genesis of bronchogenic squamous carcinoma.53
The FHIT (fragile histidine triad) tumour suppressor gene
FHIT is a tumour suppressor gene at 3p14.2, which spans the FRA3B fragile site. It appears to be a frequently deleted gene in common human carcinomas, including lung. It has been suggested that the FHIT protein might play a role in cell death through apoptosis and/or affect cell proliferation, but its true function is not known. However, its role as a tumour suppressor gene is reflected in the fact that most primary lung cancers show loss of FHIT protein expression as well as aberrant RNA and altered FHIT genome DNA.54, 55
Sozzi et al also found that FHIT protein expression is lost in 73% of non-small cell lung cancers and in 93% of precancerous lesions.56 Tumours from smokers are more likely to lose FHIT expression (75%) than those from non-smokers (39%).57 This change is independent of p53 status and seemed to be more of a feature in squamous carcinoma (87%) than in adenocarcinoma (57%). In the preinvasive lesions studied, the loss of FHIT was very frequent and appeared to increase as the grade of dysplasia increased. Moderate dysplasia showed 60% loss, whereas all cases of severe dysplasia and CIS studied showed loss of FHIT protein. Fong et al also examined preinvasive bronchial lesions, but demonstrated that 3p14.2 LOH (implying FHIT deletion) only occurred at the CIS stage and not before, suggesting that FHIT loss might be a relatively late event in central bronchial carcinogenesis.58
Genomic alterations and LOH studies
Several chromosomal sites appear to be involved frequently in lung cancer. The most common and earliest is loss of genome at 3p. In invasive disease there are at least five loci (3p12–13, 3p14.2, 3p21, 3p21.3, and 3p25) that are lost frequently. In preneoplastic lesions the identifiable losses tend to be more localised, selective, and appear relatively early. Kohno et al demonstrated that 7.7% of squamous metaplasias and 9.1% of squamous dysplasias showed LOH at 3p.45 Wistuba et al found 3p LOH more extensively, frequently and earlier, such that, in invasive squamous carcinoma and CIS, most of 3p was deleted.59 In normal or mildly abnormal epithelium, 3p loss was much less and more focal, whereas in the squamous dysplasias the degree of 3p loss was intermediate. This reflected some of the same group's earlier work, using different techniques, where a study of 3p14, 3p21.3, and 3p25 again showed early loss, with 76% of hyperplasias, 86% of dysplasias, and all examples of CIS being affected.60
The 3p loss in the early lesions did not appear to occur at random, with even the normal epithelium showing 3p21, 3p22–24, and 3p25 losses, whereas 3p14.2 and 3p14–21 losses began to appear in the hyperplastic/metaplastic lesions. These results indicate that loss of 3p14.2 occurs much earlier in the proposed sequence of events than has been suggested by Fong et al.58
Some 9p21 losses were described by Wistuba et al.59 These occurred in early disease and were almost invariably present in dysplasia, CIS, and invasive tumours. LOH at 17p13 (p53) and 13q (Rb) seemed to be a slightly later phenomenon associated with some dysplasias, but almost all CIS/invasive disease. Similarly, Kohno and colleagues45 found a 9p deletion in 9% of dysplasias and 17p LOH in 36.4%, whereas neither was shown in squamous metaplasia.
Thus, very early in the evolution of preinvasive disease, localised specific LOH of 3p and often 9p occurs, even in phenotypically normal or hyperplastic epithelium. As the lesion advances and becomes dysplastic, more 3p deletions, including 3p14.2, begin to appear and 17p LOH also becomes a feature. By the time the stage of severe dysplasia/CIS is reached, there is extensive loss of 3p and persistent loss of the 9p and 17p regions.
In a study of the next stage—early invasive squamous carcinoma (usually radiographically occult carcinoma)—Endo et al showed frequent LOH, with losses at 3p21 (53%), 5p21 (44%), and 17p13 (61%) being particularly prominent.61 Another study has shown that 5q (APC-MCC) loss might be an important event in central bronchial carcinogenesis, but not in events occurring in the periphery of the lung.62
What is not known is the relative “cause and effect” relation between these various genetic abnormalities. It is tempting to speculate that, given the relation between many of these findings and the grades of atypia (table 2), the genomic lesions or abnormal gene expression might in some way be responsible for the change in morphology. Of course the morphology of the epithelium is a mirror of genomic change, but whether or not it is a reflection of these particular changes is not known. What is known is that some of these genomic changes might be widespread in phenotypically normal epithelium, that the phenotype/histology is, in some (perhaps many) cases reversible, but when this occurs, some of the genomic alterations persist, perhaps accounting for the continuing high risk of developing lung cancer many years after the cessation of smoking. Brambilla and colleagues suggest that stable p53 overexpression, p16 loss and, perhaps, cyclin D1 overexpression in preinvasive bronchial disease might be related to a greater likelihood of progression to invasion.36, 42 These interesting findings require further attention.
The various changes occurring with progression from normal bronchial mucosa to invasive carcinoma in central bronchial carcinogenesis
So far, this discussion has primarily considered the evolution of squamous carcinoma from in situ disease in the central airway epithelium. It seems likely that some of the rare central bronchial adenocarcinomas might arise from the same preneoplastic lesion. As for small cell carcinoma, another predominantly central tumour, its progenitor lesion is essentially unknown. In a few cases, squamous dysplasia/CIS can be seen in the adjacent airway mucosa, but this is hardly proof of an origin. There has been a suggestion, based on the frequent presence of pronounced genomic abnormalities in morphologically normal mucosa near small cell tumours, that this tumour might arise almost de novo from the bronchial mucosa, without going through a stage of a morphologically recognisable preinvasive lesion.63 There are no detailed molecular biological studies on neuroendocrine cell hyperplasia.
AAH: a preinvasive lesion that progresses to adenocarcinoma
There is an increasing body of evidence to support the hypothesis that AAH is a preinvasive lesion and indeed can be regarded as the adenoma in an adenoma–carcinoma sequence in the lung periphery.11 This evidence comes from morphological observations, the association of AAH with malignancy, morphometric and cytofluorimetric data, and immunohistochemical and molecular biological studies, which are reviewed below.
MORPHOLOGICAL EVIDENCE
Most foci of atypical adenomatous hyperplasia, as described in detail earlier, look very different from lung adenocarcinoma, including BAC. However, as lesions of AAH become more atypical and larger there are no morphological features that reliably separate AAH from BAC.63 Indeed, the largest and most atypical AAH and BAC lesions might be the same thing.64 Within the scope of the current WHO definition13 of BAC as a non-invasive tumour growing by replacement of alveolar lining cells, this is easy to understand. Furthermore, most people who have studied AAH will have seen examples of (usually larger) lesions that have, for the most part, histological features of AAH but with varying grades of atypia and which, in foci, may well demonstrate invasion. The only reasonable conclusion, therefore, is that progression to invasion has occurred in part of the pre-existing AAH. Careful examination of larger well established invasive adenocarcinomas, lesions that might contain zones of BAC-like tumour, may show an AAH-like zone around the margin. Here I make the distinction (or try to) between lepidic spread of tumour and the presence of alveoli lined by cells that are much less pleomorphic than those in the tumour. Although absolute proof that these zones represent an edge of residual “adenoma” is lacking, these observations suggest such a possibility.
ASSOCIATION OF AAH WITH MALIGNANCY
Most AAH lesions are very small, cause no symptoms, and are radiologically invisible. Thus, they are found incidentally, usually in lung surgically resected for another reason. One postmortem study prospectively sought AAH and found only two patients with AAH in 100 consecutive cases.65 Given the frequency of pulmonary pathology at necropsy, such material is less than ideal for finding AAH lesions. Atypical adenomatous hyperplasia is most commonly found in lung resected for primary lung cancer, but has been found, less frequently, in lung resected for benign tumours or metastatic disease. The presence of coincident pathology such as pneumonia, obstructive pneumonitis, or fibrosis make AAH almost impossible to find. Careful examination of well inflated and formalin fixed lung (I prefer 1 cm thick parasagittal slices of resection specimens and constantly wash the cut surface of the lung with water) will improve pick up rates. Opting not to look for lesions macroscopically, relying instead on random block taking for histological identification will reduce success. Clearly, however, more blocks will help to identify more lesions. I sample all likely foci and take up to six random parenchymal blocks. Whatever the technique, determined by interest or prevailing laboratory policy, the number of AAH lesions will always be underestimated.
Overall figures for AAH incidence quoted range from 9.3% to 21.4% of cases resected from primary lung cancer, whereas lungs removed for benign or metastatic disease yield AAH in between 4.4% and 9.6% of cases.66–70 Furthermore, it is pertinent to note that AAH is more often found, and more often in large numbers, in patients who have had lung resected for adenocarcinoma or large cell undifferentiated carcinoma than for squamous carcinoma. This does appear to be a true association, although the presence of distant obstructive pneumonitis associated with a central bronchial tumour will make lesions more difficult to find. Published data show that AAH is present in up to 34.5% and 25%, respectively, of lungs resected for adenocarcinoma or large cell undifferentiated carcinoma.69 The highest published figure in squamous carcinoma is 11.1% of cases.67 Nakanishi described AAH in 6.9% of squamous cancer bearing lungs yet found lesions in 34.5% of those with adenocarcinoma.69 In our own series of 554 lungs resected for primary lung cancer, we found AAH in 12% of cases, whereas it was present in 23.2% of lungs resected for adenocarcinoma and 12.5% of large cell undifferentiated cancer resections, but in only 3.3% of lungs with squamous carcinoma.70
AAH: CASES WITH MULTIPLE LESIONS
Some patients reported in the literature have very large numbers of AAH lesions. To some extent, the number of lesions found will be proportional to the enthusiasm of the pathologist for finding them, the amount of lung tissue blocked, the presence of other pathology obscuring the lesions, the state of lung fixation and expansion, and so on. Nonetheless, it is tempting to believe that those patients with large numbers of AAH lesions might in some way be different to those with relatively few. Not infrequently, those patients with large numbers of AAH lesions also have multiple synchronous primary carcinomas, sometimes “adenocarcinoma in AAH”. Those patients with many AAH lesions almost invariably have adenocarcinoma as their associated malignancy.
The patient described by Nadav and colleagues71 had three synchronous primary BAC-type adenocarcinomas (but a single AAH lesion) associated with Li-Fraumeni syndrome, whereas two of Weng and colleague's patients had multiple AAH and two synchronous primary lung cancers.66 Suzuki et al describe a patient with bilateral stage I BAC lesions and also 12 AAH lesions.72 Patients with LOH in the tuberous sclerosis gene associated regions in adenocarcinoma may have multiple AAH lesions.73 Anami et al also described a patient who underwent left upper lobectomy; the operative specimen yielded two papillary adenocarcinomas and 161 AAH lesions.74
In our series of 70 patients with AAH, eight patients had between six and eight AAH lesions, whereas four had 12, 19, 34, and 42 lesions. Of these patients all had adenocarcinoma, except one who had a mixed carcinoma, including adenocarcinoma. Ten of the 70 patients had multiple synchronous primary adenocarcinomas (up to six in each resection specimen) and many had multiple AAH lesions.70
It is not known whether patients with such a propensity to develop large numbers of AAH lesions and cancers have a particular constitutive genetic lesion underlying this change. The patient described above with Li-Fraumeni syndrome had only one AAH lesion. Nonetheless, this will be an interesting area of future study.
AAH: NUCLEAR SIZE, DNA CONTENT, AND CELL PROLIFERATION
In 1986 Kodama et al morphometrically measured mean nuclear area (MNA) and demonstrated an increase in mean values from low, through high grade AAH, to adenocarcinoma.75 Also of interest was their finding of two cell populations in two lesions. In each of these lesions (one was (apparently) an AAH, the other an adenocarcinoma), one cell population had a scatter of MNA typical of adenocarcinoma, the other was characteristic of AAH. This is of interest given the morphological observations referred to above. A similar progression of increasing MNA from AAH through BAC to “overt BAC” (adenocarcinoma with bronchioloalveolar features) was described by Kitamura et al.76
Nuclear DNA content was measured cytofluorimetrically in a range of AAH lesions and small adenocarcinomas by Nakayama et al.77 Using this measure, small solitary AAH lesions showed less DNA than areas of AAH in conjunction with adenocarcinoma, whereas the data for this latter group were indistinguishable from those for adenocarcinoma. They also found aneuploid stem cell lines in 53.8% of AAH foci and 76.9% of adenocarcinomas. These authors conclude that AAH was either an adenoma or a well differentiated adenocarcinoma.
In a more sophisticated morphometric study using 12 variate cluster analysis, Mori and colleagues78 found that lesions classified morphologically as AAH could be separated into two groups, one of which showed characteristics identical to adenocarcinomas of type II pneumocytes. These AAH lesions tended to be more atypical than those without an adenocarcinoma-like data pattern.
Argyrophilic nucleolar organiser region (AgNOR) counts were shown to increase from low grade to high grade AAH and to BAC by Nakanishi et al.79 They also examined DNA aneuploidy and found none in low grade AAH, but detected aneuploidy in 25% of high grade AAH and 34.6% of BAC lesions. There are few studies of cell proliferation in AAH but ours and others, using anti-PCNA markers, found that the growth fraction in AAH is very low.80–82
All of these findings provide more objective evidence for the morphological observations of increasing atypia in AAH and the transition to adenocarcinoma.
AAH: IMMUNOHISTOCHEMISTRY
Several antigens have been shown to be expressed, or overexpressed, in AAH when compared with normal peripheral lung epithelium. Many authors have shown the expression of carcinoembryonic antigen (CEA), with a tendency for more to be expressed in larger or more atypical lesions.67, 69, 76, 83 CEA is almost universally expressed in lung adenocarcinoma.
Mori et al,84 in a re-examination of cases subjected to detailed morphological analysis, studied low and high grade AAH and BAC with a range of antibodies, including anti-CEA. Other antigens stained for were surfactant apoprotein A (SAP), urine protein 1 (UP1), and cytochrome p450. These authors reinforced the conclusion of their earlier work—that high grade AAH lesions are closely related to BAC—by finding that both high grade AAHs and BACs had identical staining profiles (high CEA, p450 (including CYP1A1), and SAP and low UP1), whereas low grade AAH lesions showed the reverse. A later study by Kitamura and colleagues85 also used antibodies to UP1 and SAP, but found that UP1 was not expressed in AAH lesions but was found in BAC and was widespread in “overt BAC” (adenocarcinoma with bronchioloalveolar features). SAP staining was found in all lesions but appeared to decrease as the lesions advanced.
Blood group antigens expressed in normal alveolar epithelium and low grade AAH are also lost as lesions progress to high grade atypia and BAC.69
AAH: ONCOGENES AND TUMOUR SUPPRESSOR GENES
One of the earliest studies of oncogene expression in AAH lesions stained for p53 protein and c-erbB2, the product of the neu oncogene.86 Only 7% of AAH lesions expressed c-erbB2, and these were universally high grade lesions. Expression of p53 protein was found in 58% of all lesions, with higher grade AAH lesions showing more staining. Other studies have found either similar staining,87 or much less stainable p53 protein in 10% of AAH lesions,76 although the relation with AAH grade was preserved. In a small series of patients with cancer, Katabami and colleagues41 found that although 25% of AAH lesions in patients without pneumoconiosis had raised p53, 36.4% of AAH lesions in patients with pneumoconiosis showed excess staining.
Slebos et al found stainable p53 in none of their low grade AAH lesions, 9% of high grade AAH, and in 50% of their early BAC samples.88 One high grade AAH lesion showing p53 protein overexpression had a mutation in the p53 gene, whereas three others showed no mutation. Kohno and colleagues45 also found p53 gene mutation to be an infrequent event, occurring in only one lesion of 20 examined, and concluded that p53 mutation may be a relatively late event in peripheral adenocarcinogenesis, when compared with events leading to the development of bronchial squamous carcinoma.
In a study comparing p53 expression with the expression of p53 inducible cyclin dependent kinase inhibitor p21Waf1/Cip1, Hayashi et al found that, although reactive lesions showed a correlation between p53 and p21 expression, this was lost in both AAH and carcinomas of various stages.80 They suggested that p21 expression might become a p53 independent event during neoplastic transformation, even at the very early AAH stage.
Cell cycle regulators were studied by Kurasono et al.81 They found that, whereas cyclin D1 appeared to be overexpressed in most AAH foci, but less so in BAC, the loss of Rb and p16 expression was rare or not seen at all in AAH, and was relatively infrequent in adenocarcinoma. They suggested that although cyclin D1 overexpression might occur early in adenocarcinoma tumorigenesis, it did not appear to be required to maintain the malignant phenotype.
Four studies have reported the presence of K-ras mutation (point mutation at codon 12) in AAH. This is an important finding because K-ras mutations are frequent in human pulmonary adenocarcinoma. Such mutations were first described by Ohshima et al in 1994,89 who found mutation in AAH in two of six patients studied. Sagawa et al found only one AAH lesion with a K-ras mutation in the 39 lesions examined.90 Westra et al found that 39% of AAH lesions had the mutation, which was evenly distributed among different grades of AAH and similar in frequency to that found in adenocarcinoma.91 Furthermore, these authors showed that the AAH lesion often showed different base changes at codon 12 of the K-ras gene when compared with those found in the concurrent adenocarcinoma from the same patient. This is consistent with the notion that AAH represents “field cancerisation” in the lung periphery. In our own work51 K-ras mutation was found in fewer AAH lesions (15% of those studied) but was seen in 35% of peripheral parenchymal type adenocarcinomas (adenocarcinoma with bronchioloalveolar features), the type of adenocarcinoma arising from AAH, although never being found in adenocarcinomas of bronchial origin.
AAH: LOSS OF HETEROZYGOSITY AND OTHER GENOMIC ALTERATIONS
Much work, already detailed above, has been carried out on the preinvasive lesions that occur in the central bronchial epithelium. Much less is known of AAH lesions. Kohno et al studied 28 AAH lesions, eight low grade and 20 high grade.45 No LOH was identified in low grade lesions but two high grade lesions showed 3p LOH and one showed 9p loss. Although no AAH lesions were studied by Suzuki et al,73 they showed that lungs with an adenocarcinoma, which demonstrated LOH in the tuberous sclerosis gene associated regions (TSC1 and TSC2–9q and 16p, respectively), were more likely also to have multiple AAH lesions. Nadav et al also described an interesting patient with Li-Fraumeni syndrome who had multiple synchronous lung cancer and AAH lesions.71 Anami et al studied a single unusual patient who had two synchronous lung adenocarcinomas and 161 AAH lesions.74 Six AAH lesions greater than 3 mm were examined and 17q LOH was shown in two, 9q LOH in one, whereas another demonstrated microsatellite instability at D17S791. Kitaguchi et al studied 31 AAH lesions and showed 3p, 9p, and 17p loss in 19%, 13%, and 5%, respectively.92 All lesions with genetic alterations were higher grade. The 3p LOH is clearly of interest, even though to date very few lesions have been studied, given the finding that 43% of BAC lesions show evidence of LOH within the FHIT gene at region 3p14.2.93
AAH lesions have been shown to be monoclonal proliferations by analysis based on the X-linked polymorphic human androgen receptor gene.94 Of interest also, in this paper, was the presence of identical monoclonality in two cases of BAC and the contiguous AAH. This finding supports the earlier contention that morphological observations fit with the hypothesis that the AAH-like edge, which is sometimes observed around a peripheral adenocarcinoma (with or without bronchioloalveolar features), is evidence of the pre-existing adenoma from which the invasive carcinoma arose.
AAH: FATE OF THE LESION
Despite the increasing amount of data, summarised above and in table 3, which support a progression towards malignancy at a morphological and molecular level, there are no data to suggest the risk of progression of AAH to invasive carcinoma. The almost impossible task of identifying these lesions before resection rules out any longitudinal study. It remains to be seen whether or not state of the art spiral computed tomography (CT) scanning can offer some chance of detecting the largest examples of AAH.95, 96
The various morphological, morphometric, immunohistochemical, and molecular biological findings in AAH and adenocarcinoma
In 1997 Suzuki et al described a follow up study where patients, whose resected cancers were associated with AAH, were compared with those who did not have AAH.97 The presence of AAH in the lungs did not appear to affect the survival of the patients adversely. Conversely, in another study98 there is evidence that patients with stage IA disease and AAH had a better postoperative survival than those without AAH. Our data mirror those of Suzuki et al.70
Morphological observations of our own cases suggest that a small proportion of AAH lesions might undergo sclerosis and possible regression, leaving behind a scar in the lung, which retains the topography of an AAH lesion. Sometimes this process affects only part of the lesion, and some or all of the patient's lesions may show this change. What relation this has with the heavy infiltration of AAH by lymphocytes occasionally seen in some patients is not known.
Until it becomes possible to make longitudinal observations of lesions, or an animal model is found, evidence that AAH does progress through BAC to invasive adenocarcinoma will largely be based on observational and circumstantial evidence, as described in this review.
Pulmonary preinvasive lesions: relevance to clinical practice
Apart from academic interest in the process of carcinogenesis, a knowledge of pulmonary preinvasive lesions also has relevance to several clinical situations. If progress is ever to be made with screening for lung cancer, then more needs to be understood about its progenitor lesions. It is clear that detecting pulmonary neoplasia at a stage when invasion is already present is too late. To date, screening has largely been based upon examining sputum specimens for atypical cells, or mass radiography campaigns. Morphology on sputum alone is not good at differentiating invasive from preinvasive bronchial disease, and it remains to be seen whether any other specific marker can be found that can reliably identify exfoliated neoplastic cells. However, fluorescence bronchoscopy (LIFE) might increase the detection of squamous dysplasia/CIS, a lesion more or less invisible using standard visualisation methods. Histopathologists may face having to deal with large numbers of biopsies from bronchi examined using the LIFE technique. The experience of Lam et al would suggest that among the frequent non-atypical biopsy specimens taken during LIFE procedures, a variety of dysplasias will be diagnosed.14 What then? We have so little idea of how these progress and currently have such limited therapeutic options that these diagnoses may be of limited clinical value. Their presence might help to persuade a patient to stop smoking, and lesions may be treatable by photodynamic therapy, but the real hope is that some form of chemopreventive intervention can be offered, with surveillance using fluorescence bronchoscopy. Will this be yet another contribution to the ever increasing numbers of biopsy specimens pouring through the doors of histopathology departments?
The lung periphery is much harder to “get at”. Unless a very specific marker comes to light, I doubt whether bronchoalveolar lavage has anything to offer in terms of detecting AAH. Given the size and architecture of most lesions, they are invisible on plain chest x ray films or CT scans using standard resolution. Even high resolution CT would be unlikely to produce a specific enough appearance to be diagnostic. Nonetheless, the work of Eguchi and colleagues95 and Sone and colleagues96 is of interest because leading edge radiology might provide a means of detecting relatively large lesions that could be excised, perhaps using stereotactic needle localisation. This again, opens the door to a potential screening situation. This prospect, in turn, raises the issue of the diagnosis of AAH and its differentiation from BAC, as currently defined by the WHO. Using existing criteria, distinguishing a high grade AAH from BAC is very difficult, certainly using morphology alone. As yet, no reliable adjunct marker or group of markers has been found to help. In any case, it may be pointless to try to separate what is essentially the same disease, pulmonary alveolar intraepithelial neoplasia or adenocarcinoma in situ.
Acknowledgments
Thanks to Dr A Chapman, Mr S Pritchard, and Professor J Simpson for their review and comments on this manuscript.