


Abstract
The exact role of p38 mitogen-activated protein kinase (MAPK) in the expression of inflammatory cytokines is not clear; it may regulate transcriptionally, post-transcriptionally, translationally, or post-translationally. The involvement of one or more of these mechanisms has been suggested to depend on the particular cytokine, the cell type studied, and the specific stimulus used. Interpretation of some of the published data is further complicated by the use of inhibitors such as 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (SB 203580) used at single, high concentrations. The aim of this study was to determine the impact of two second-generation p38 MAPK inhibitors on the expression of a range of inflammatory cytokines at the gene and protein levels in human cultured cells. Similar assessment of the impact of these compounds on inflammatory cytokine expression in a preclinical in vivo model of airway inflammation was performed. The results in THP-1 cells and primary airway macrophages clearly show that protein expression is inhibited at much lower concentrations of inhibitor than are needed to impact on gene expression. In the rodent model, both compounds, at doses that cause maximal inhibition of cellular recruitment, inhibit tumor necrosis factor α (TNFα) protein production without impacting on nuclear factor κB pathway activation or TNFα gene expression. In summary, the data shown here demonstrate that, although at high compound concentrations there is some level of transcriptional regulation, the predominant role of p38 MAPK in cytokine production is at the translational level. These data question whether the effect of p38 inhibitors on gene transcription is related to their potential therapeutic role as anti-inflammatory compounds.
The mitogen-activated protein kinase (MAPK) signal transduction pathway is believed to play an integral role in the innate response to invading pathogens. Of the three main MAPKs, p38 is believed to be involved in the production of cytokines and chemokines vital for the recruitment of effector white cells central to the innate immune response, such as neutrophils. Inhibition of p38 MAPK, by compounds such as SB 203580, has been shown to block lipopolysaccharide (LPS)-induced production of cytokines, such as TNFα and IL-8, from monocytes/macrophages (Young et al., 1993; Lee et al., 1994; Manthey et al., 1998; Nick et al., 2000). In preclinical rodent models of LPS-driven airway inflammation, studies have shown a reduction of cytokine production and inflammatory cellular recruitment in the presence of p38 MAPK inhibitors (Nick et al., 2000, 2002; Underwood et al., 2000a,b; Haddad et al., 2001). The exact role of p38 MAPK in the expression of inflammatory cytokines is not clear; a recent review by Newton and Holden (2003) collates data indicating that this kinase may regulate transcriptionally, post-transcriptionally, translationally, or post-translationally. Indeed, the review suggests that the involvement of one or more of these mechanisms can depend on the particular cytokine, the cell type studied, and the stimulus used. Initial studies, with inhibitors such as SB 203580, indicated that p38 MAPK was exclusively involved in the translational control of cytokine expression (Young et al., 1993; Lee et al., 1994; Kotlyarov et al., 1999). However, other publications have suggested a role of p38 MAPK in the regulation of several proinflammatory transcription factors such as activating transcription factor-2 (Chen et al., 1998), NF-κB (Raingeaud et al., 1996; Nick et al, 1999), and, indirectly, activator protein-1 (Newton and Holden, 2003). In addition, p38 MAPK has been reported to indirectly impact on the NF-κB pathway by regulating chromatin modulation, affecting the access to DNA binding sites (Saccani et al., 2001). p38 MAPK has also been shown to increase the stability of mRNA for genes containing adenosine/uridine-rich elements (Wang et al., 1999; Brook et al., 2000).
Although it does seem that the role of p38 MAPK in expression of cytokines is complex, interpretation of some of the published data is further complicated by the use of inhibitors such as SB 203580 at single, high concentrations. The aim of this study was to determine the impact of two second-generation p38 MAPK inhibitors, SB 239063 (reported activity at the p38α ATP binding site is 50 nM; Underwood et al., 2000a,b) and compound 2 (reported activity at the p38α ATP binding site is 40 nM), on the expression of a range of inflammatory cytokines at the gene and protein levels. Activity (IC50 in nanomoles) of SB 239063 and compound 2 on other kinases was reported to be >10,000 and 2500 for c-Jun N-terminal kinase-1; >10,000 and >10,000 for Iκ-B kinase-2; >10,000 and >10,000 for ROCK; >10,000 and >10,000 for AKT; >10,000 and >10,000 for Alk5; 2000 and >10,000 for Lck; >10,000 and >10,000 for raf-MEK-ERK, respectively, using assays based on methodologies published in Lee et al. (1999). Concentration-response curves were performed in LPS-stimulated human monocyte cells and primary cultured human lung tissue macrophages, which allowed comparison of the impact at the gene and protein levels across a range of compound exposure levels. These two inhibitors were also profiled in a preclinical rodent model of LPS-induced airway inflammation, in which we determined the effect on cytokine gene and protein expression as well as the impact on innate effector cell recruitment in vivo.
Materials and Methods
Culture of THP-1 Monocytic Cells
The human monocytic cell line THP-1 was purchased from the European Collection of Cell Cultures (ECACC; Centre for Applied Microbiology and Research, Salisbury, Wiltshire, UK). Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 with glutamax I (Invitrogen LTD, Paisley, UK) supplemented with 10% fetal calf serum and 1% antibiotic and antimycotic solution (penicillin/streptomycin; Sigma-Aldrich Co., Poole, UK) at 37°C in a humidified atmosphere [95% air, 5% (v/v) CO2]. The cells were cultured into 75-cm2 flasks, and the media were replaced after 3 days and thereafter every 48 h. The media were changed by centrifuging the supernatant at 800g for 5 min at room temperature in a chillspin (Mistral 3000i; MSE, London, UK). The supernatant was discarded, and the pellet of cells was resuspended in 1 ml of RPMI 1640 with glutamax I, supplemented with 10% fetal calf serum and 1% antibiotic and antimycotic solution. A trypan blue cell count was performed, and cells were passaged into 2 × 75-cm2 flasks when cell numbers reached 10 × 106 cells/ml. This cell line has a doubling time of approximately 48 h. On experimental days, the cells (10 × 106 cells/ml) were centrifuged at 800g for 5 min at room temperature in a chillspin (Mistral 3000i; MSE). The supernatant was discarded, and the pellet of cells was washed in 10 ml of RPMI 1640 with glutamax I (Invitrogen LTD) supplemented with 1% antibiotic and antimycotic solution (penicillin/streptomycin; Sigma-Aldrich Co.) with no fetal calf serum. The cells were then centrifuged under the same conditions, the supernatant was discarded, and the cells were resuspended in RPMI 1640 with glutamax I, supplemented with 3% fetal calf serum and 1% antibiotic and antimycotic solution.
Extraction of Human Lung Tissue Macrophages
Human lung tissue macrophages were obtained from nondiseased donor tissue that was not suitable for transplant as outlined below. Ethical approval for the study was obtained along with consent from the relatives. Lung tissue was cut into small pieces and flushed with phosphate-buffered saline using a needle and syringe (without calcium and magnesium) (Sigma-Aldrich Co.). The pooled cell suspension was passed through a 70-μm cell sieve and centrifuged at 250g for 10 min at 4°C in a chillspin (Mistral 3000i; MSE). The supernatant was discarded, and the cell pellets were resuspended in phosphate-buffered saline (without calcium and magnesium) and layered onto six discontinuous Percoll gradients [60%/35%/25% (v/v)]. These gradients were then centrifuged at 1200g for 25 min at 20°C. After centrifugation, the macrophage enriched fractions were obtained from the 35% and the 60% Percoll interface and washed twice with phosphate-buffered saline. The cells were then resuspended with RPMI 1640 with glutamax I (Invitrogen LTD), supplemented with 10% fetal calf serum (Invitrogen LTD) and 1% antibiotic and antimycotic solution (penicillin/streptomycin; Sigma-Aldrich Co.). Trypan blue exclusion was performed to assess cell viability, and cell purity of the macrophage-enriched fraction was determined with Kimura stain. The cell suspension was diluted in RPMI 1640 with glutamax I, supplemented with 10% fetal calf serum and 1% antibiotic and antimycotic solution, and 400,000 cells per well were added to 24-well plates (Costar). These plates were then incubated for 60 min at 37°C in a humidified atmosphere [95% air, 5% (v/v) CO2]. After 60 min, the nonadherent cells were removed by washing, and fresh medium was added. The adherent, purified macrophages (consistently >99%) were incubated overnight for treatment the following day.
Compound Treatment of THP-1 Monocytic Cells and Human Lung Tissue Macrophages
For analysis of supernatant for protein expression, 225 μl of THP-1 cell suspension containing 400,000 cells were seeded onto 24-well plates, whereas for RNA extractions or nuclear extractions, 900 μl of THP-1 cell suspension containing 1.6 × 106 cells were seeded onto six-well plates. For macrophages, either 225 or 900 μlof RPMI 1640 with glutamax I, supplemented with 3% fetal calf serum and 1% antibiotic and antimycotic solution, was added to the 24- or six-well plates of adherent cells, respectively. Varying concentrations (THP-1, 0.1 nM–1 μM; macrophages, 10 nM–10 μM) of SB 239063 and compound 2 (a kind gift from GlaxoSmithKline, Uxbridge, Middlesex, UK) were assessed in this study, and dexamethasone (1 μM) was used as the positive control. Two-hundred fifty microliters (for 24-well plates) or 1000 μl (for six-well plates) of each concentration of drug was added to the designated wells and incubated for 60 min at 37°C in a humidified atmosphere [95% air, 5% (v/v) CO2]. After 60 min, 25 μl (for 24-well plates) or 100 μl (for six-well plates) of vehicle [0.01% (v/v) phosphate-buffered saline (PBS)] or LPS (0.1 μg/ml) was added, and the plates were then incubated at 37°C in a humidified atmosphere [95% air, 5% (v/v) CO2] for 2 h (for gene expression and extractions) or 24 h (for protein expression). After 2 and 24 h, THP-1 cell suspensions were transferred from the plates into centrifuge tubes and were centrifuged at 800g for 5 min at 4°C in the Mistral chillspin. Supernatants were retained for protein expression by enzyme-linked immunosorbent assay (ELISA), and the cell pellets were retained for either gene expression or total protein extract. For plates of adherent macrophages, the supernatants were also retained for protein expression by ELISA, and the plates of adherent cells were frozen for either gene expression or total protein extract.
Determination of the Effect of SB 239063 and Compound 2 on LPS-Induced Cytokine (Protein) Production from Cultured THP-1 Cells and from Human Lung Tissue Macrophages
Levels of TNFα, IL-8, IL-6, MIP-1α, GROα, GM-CSF, and G-CSF in the supernatant retained from THP-1 cells and human lung tissue macrophages were determined by ELISA using human Duosets according to the manufacturer's instructions (R&D Systems Europe, Oxfordshire, UK).
Determination of the Impact of SB 239063 and Compound 2 on LPS-Induced Cytokine (Gene) Production from Cultured THP-1 Cells and from Human Lung Tissue Macrophages
RNA Extraction and Reverse Transcription. Total cellular RNA was isolated from THP-1 cells and human lung tissue macrophages using Tri Reagent (Sigma, Poole, UK). Briefly, 1 ml of Tri Reagent was added to each pellet of THP-1 cells and to each well of a six-well plate of adherent macrophages, which were then transferred to centrifuge tubes, and manufacturer's instructions were followed to extract RNA from the cells. The purity and integrity of the RNA samples was assessed by A260/A280 spectrophotometric measurements on the GeneQuant RNA/DNA quantifier (Amersham Pharmacia Biotech, UK).
RNA samples (1 μg) were reverse transcribed using a master mix (TaqMan reverse transcription reagents; Applied Biosystems, Warrington, Cheshire, UK) containing 1× TaqMan RT buffer, 5.5 mM MgCl2, deoxyNTPs mixture (500 μM per NTP), 2.5 μM random hexamers, 0.4 U/μl RNase inhibitor, and 1.25 U/μl Multiscribe reverse transcriptase in a final reaction volume of 50 μl. The RNA samples were incubated in a Perkin-Elmer 480 thermal cycler (Boston, MA) at 25°C for 10 min, then reverse transcribed at 48°C for 30 min, and finally, the enzyme was denatured at 95°C for 5 min. Samples were then stored at –80°C until required for analysis.
Cytokine mRNA Expression. Transcriptional expression of target mRNA transcripts in RNA samples were detected by polymerase chain reaction (PCR) amplification and quantified by 5′-nuclease assay using fluorescent-labeled TaqMan probes (TaqMan; Applied Biosystems) and analyzed using TaqMan real-time quantitative PCR with the ABI PRISM 7700 Sequence Detection System (Applied Biosystems).
Predeveloped assays (Assay on Demand) were purchased from Applied Biosystems, for the investigation of gene expression of target genes (TNFα, IL-8, IL-6, GROα, and MIP-1α) in THP-1 cells and human lung tissue macrophages. Reactions were internally controlled with the 18s rRNA internal control (Applied Biosystems). PCR reactions were performed in a 25-μl reaction volume containing 3 μl of sample cDNA (2.5 ng/μl), with the Assay on Demand of the target gene, 2× TaqMan Universal Master Mix and 18s internal control (Applied Biosystems).
Amplification and detection of specific products were carried out in an ABI PRISM 7000 sequence detection system (Applied Biosystems) using an amplification protocol consisting of 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. Results were analyzed using the Sequence Detection Software (Applied Biosystems), and the relative amount of target gene transcript was normalized to the amount of 18S internal control transcript in the same cDNA sample. The data were then compared with levels in the vehicle control group and are presented as -fold increase over control alone.
Effect of Compounds on a Downstream Target for p38 MAPK and a Marker of Translation: Mnk1 Phosphorylation. To demonstrate that the two p38 MAPK inhibitors actually impact on the kinase and inhibit inflammatory mediator protein translation, levels of phosphorylated Mnk1 was measured. Mnk1 has been shown to be phosphorylated by p38 MAPK, and phosphorylation of this protein is critical in the translational process (Pyronnet et al., 1999; Waskiewicz et al., 1999).
THP-1 cells were cultured as above, except the experiments were performed in six-well plates (1.6 × 106 cells per well) and treated with either compound (1 nM–1 μM) for 1 h and then exposed to LPS (0.1 μg/ml). Two hours after stimulation (a time point previously established to be appropriate for demonstrating an increase in phosphorylated Mnk1), the cell pellets were collected. Total protein was extracted with 0.5 ml of ice-cold lysis buffer (50 mM Tris-HCl, pH 6.8, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, and 0.01 M EDTA) containing protease and phosphatase inhibitors (25 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 5 mM dithiothreitol, 0.5 mM phenylmethyl sulfonyl fluoride, 2 mM sodium orthovanadate, 1.25 mM sodium fluoride, and 1 mM sodium pyrophosphate). Cell debris was pelleted by centrifugation, and the supernatants were removed and stored. Western blot analysis of proteins was carried out using the NuPAGE system (Invitrogen) per the manufacturer's instructions. All samples were run on 4 to 12% gradient gels in MOPS buffer supplemented with antioxidant (Invitrogen). Proteins were electrotransferred onto nitrocellulose membranes using the NuPAGE transfer buffer. Detection of proteins was performed using enhanced chemiluminescence (Amersham Pharmacia Biotech, Little Chalfont, Bucks, UK) according to the manufacturer's instructions. The primary antibody used was anti-phospho-Mnk1 (Thr197/202, 1:1000; Cell Signaling Technology Inc.), and the secondary antibody was anti-rabbit immunoglobulin/horseradish peroxidase (1:2500; DakoCytomation Ltd.). Housekeeping protein used was actin (1:1000; Sigma-Aldrich Co.).
Determination of the Effect of SB 239063 and Compound 2 on LPS-Induced Airway Inflammation in the Rat
Male Wistar rats (150–180 g) were purchased from Harlan-Olac (Bicester, UK) and housed for at least 5 days before use. Food and water were supplied ad libitum. UK Home Office guidelines for animal welfare based on the Animals (Scientific Procedures) Act 1986 were strictly observed.
Rats were challenged with an aerosol of endotoxin-free saline (for 30 min) or LPS (0.3 mg/ml) as previously described (Haddad et al., 2001). Vehicle (0.5% methylcellulose and 0.2% Tween 80 in water, 2 ml/kg), SB 239063 (3, 10, or 30 mg/kg), or compound 2 (1, 3, 10, or 30 mg/kg) was administered orally (p.o.) 1 h prior to and 2 h postchallenge. The positive standard dexamethasone (0.01, 0.1, or 1 mg/kg) was included and administered using the same treatment regimen. Six hours after challenge, cellular inflammation, myeloperoxidase (MPO) activity, NF-κB p65 DNA binding, and TNFα and IL-1β gene expression and protein levels were determined using methodology detailed in Birrell et al. (2005). Briefly, animals were euthanized with sodium pentobarbitone (200 mg/kg i.p.), and the airways were lavaged with RPMI (1 ml/100 g of body weight times 2, pooled), the left lobe was removed and the vasculature perfused with RPMI to remove any blood and then finely chopped. A 300-mg aliquot of the lung tissue went through an enzymatic digest to extract the white cells. The remaining lung was flash-frozen in liquid nitrogen and stored at –80°C for analysis of cytokine protein and gene expression.
Total white blood cell counts in the lung samples were determined using an automated cell counter (Sysmex F-820; Sysmex UK Ltd., Linford Wood, Buckinghamshire, UK). Cytospins of these samples were prepared by centrifugation of 100-μl aliquots in a cytospin (Shandon, Runcorn, UK) at 700 rpm for 5 min, low acceleration at room temperature. Slides were fixed and stained on a Hema-tek 2000 (Ames Co., Elkhart, IL) with modified Wrights-Giemsa stain. Four-part differential counts on 200 cells per slide were performed following standard morphological criteria, and the percentage of eosinophils, lymphomononuclear cells, and neutrophils were determined.
For the determination of MPO levels in the lung tissue, approximately 200 mg of lung tissue was homogenized in 2 ml of ice-cold saline using an Ultraturrax T25 homogenizer. The samples were then spun in a benchtop microcentrifuge at 13,000g for 10 min [MSE Micro Centaur; Jencons (Scientific) Ltd., Leighton Buzzard, Bedfordshire, UK].
MPO content was determined in lung homogenates using a spectrophotometric reaction with o-dianisidine hydrochloride as a substrate. Briefly, 240 μl of substrate solution (50 mM phosphate, 0.5% hexadecyltrimethylammonium buffer containing 0.167 mg/ml o-dianisidine hydrochloride), 10 μl of H2O2 (0.0005%), and 10 μl of sample or standards (human MPO from Sigma tested at 0, 0.078, 0.156, 0.3125, 0.625, 1.25, 2.5, and 5 U/ml) were mixed. The addition of sample starts the reaction. Absorbance was read at 450 nm on a plate reader, and the results expressed as units per milligram of total protein. MPO levels were corrected for total protein content, which was measured using the Bradford assay.
Cytokine levels in the bronchoalveolar lavages were determined by ELISA using commercially available kits according to manufacturer's instructions. TNFα and IL-1β were determined using a rat-specific sandwich immunoassay kit obtained from R&D Systems, Inc. (Minneapolis, MN).
NF-κB activity was measured using a TransAM NF-κB p65 ELISA-based assay kit (Activ Motif, Rixensart, Belgium). Briefly, lung tissue was homogenized in lysis buffer (20 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, 0.1% Nonidet P40, 0.05% sodium deoxycholate, 0.025% SDS, and 0.1% Triton X-100) containing 5 mM dithiothreitol and complete EDTA-free protease inhibitors (one tablet in 50 ml of buffer). Protein concentration was determined by Bradford assay, and 100 μg of protein from each sample was used in the assay. Samples (20 μl) were placed along with 70 μl of binding buffer on a 96-well plate, to which oligonucleotide containing an NF-κB consensus binding site had been immobilized, for 1 h on a rocker. During this time, the activated NF-κB contained in the sample specifically binds to this nucleotide, then the plate was washed, and by using a primary antibody (100 μl diluted 1:1000 in antibody binding buffer for 1 h) that is directed against the NF-κB p65 subunit, the NF-κB complex bound to the oligonucleotide is detected. The plate was then washed again, and 100 μl of secondary antibody (diluted 1:1000 in antibody binding buffer) conjugated to horseradish peroxidase was added for 1 h. The plate was washed again, and 100 μl of developing solution was added. The plate was incubated for 2 to 10 min away from direct light; 100 μl of stop solution was added, and the plate was read using the MCC/340 Multiskan plate-reader at 450 nm.
Extraction of total cell RNA, reverse transcription, and gene expression in the lung tissue was determined following a similar method outlined above for the cell-based assays. TNFα mRNA expression was measured using predetermined assay reagents purchased from Applied Biosystems. To demonstrate that the two compounds impacted on a downstream target for p38 MAPK and a marker of protein translation, Western blot analyses were performed for phosphorylated Mnk1 from homogenized lung tissue using the methods described above.
Materials
SB 239063 and compound 2 were kind gifts from GlaxoSmithKline. LPS from Escherichia coli serotype 0111:B4 was purchased from Sigma. Roche Diagnostics (Lewes, UK) supplied the DNase and collagenases. RPMI 1640 medium, HEPES, glutamax, PBS, and fetal calf serum were all obtained from Invitrogen. All ELISA Duoset kits were from R&D Systems (Abingdon, UK). All reverse transcription-PCR reagents and Assay on Demands were obtained from Applied Biosystems (Warrington, UK). Sodium pentobarbitone (Euthatal) was from National Veterinary Supplies (Harlow, UK).
Data Analysis
All the values in the figures and text are expressed as mean ± S.E. mean of n observations. Impacts of LPS exposure were compared using Student's t test with a Mann-Whitney U post test for unpaired data. Statistical analyses of multiple comparisons were made using the Kruskal-Wallis test followed by a Dunn's post test. All treatments were compared with relevant vehicle control groups; differences were deemed significant when P < 0.05.
Results
Determination of the Effect of SB 239063 and Compound 2 on LPS-Induced Cytokine (Protein) Production from Cultured THP-1 Cells and from Human Lung Tissue Macrophages. LPS stimulation caused a significant increase in TNFα, IL-8, GROα, and MIP-1α protein levels from THP-1 cells (Figs. 1 and 2). Pretreatment with SB 239063 and compound 2 significantly inhibited the LPS-induced release of all four cytokines with approximate IC50 values of 10 nM (Figs. 1 and 2). LPS stimulation of the human lung tissue macrophages caused an increased protein expression of TNFα, IL-8, IL-6, MIP-1α, GM-CSF, and G-CSF (Figs. 3 and 4). The two p38 MAPK inhibitors caused a concentration-related decrease in cytokine expression similar to the impact seen on THP-1 cells; however, the magnitude of the inhibition appeared less, especially on certain cytokines, i.e., IL-8, and the potency of the compounds appeared 10-fold lower.
Determination of the Effect of SB 239063 and Compound 2 on LPS-Induced Cytokine (Gene) Production from Cultured THP-1 Cells and from Primary Human Lung Macrophages. LPS stimulation of THP-1 cells caused a significant increase in cytokine gene expression (Figs. 5 and 6). Pretreatment with either of the compounds had no significant effect on TNFα, GROα, and MIP-1α gene expression, although in some cases there was some reduction at the highest concentration used (Figs. 5 and 6). Interestingly, both compounds inhibited LPS-induced IL-8 gene expression, which reached statistical significance with compound 2 (1 μM) and significantly inhibited basal IL-8 gene expression.
For examination of the impact of these compounds on gene expression in macrophages, we measured levels of IL-6 as the protein expression of this cytokine was affected by the p38 MAPK inhibitors to the greatest extent. Expressed as -fold difference from basal levels pretreatment with SB 239063 and compound 2 (0.01, 0.1, 1, and 10 μM) resulted in values of 26.6, 27.1, 27.4, and 16.1 compared with a vehicle of 28.9 and 29.5, 30.2, 27.4, and 13.8 compared with a vehicle of 24.0, respectively. These data demonstrate that, similar to the results obtained with the THP-1 cells, neither compound impacted on gene expression at concentrations, which inhibited protein expression. Although there was inhibition of gene expression at the highest concentration used, 10 μM.
Demonstration of the Activity of the Two p38 MAPK Inhibitors in the Human Cell-Based Assay System and That They Impact at the Level of Protein Translation. Stimulation with LPS caused an increase in the levels of phosphorylation of Mnk1 in the cultured human cells (Fig. 7). Preincubation with the two p38 MAPK inhibitors caused a concentration-related inhibition of this increase phospho-Mnk1, which appeared to directly correlate with the inhibition of inflammatory proteins observed (Figs. 1, 2, and 7).

Effect of SB 239063 and dexamethasone on cytokine protein expression from LPS-stimulated THP-1 cells. THP-1 cells were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], SB 239063 (0.1 nM to 1 μM), or dexamethasone (1 μM) before stimulation with vehicle [0.01% (v/v) PBS] or LPS (0.1 μg/ml). Twenty-four hours later, the cell culture supernatants were collected and stored at –80°C until required for cytokine assessment by ELISA (A, TNFα; B, IL-8; C, GROα; and D, MIP-1α). Results are expressed as mean ± S.E. mean of three experiments run in duplicate. Statistical significance between groups was assessed using Student's t test (two groups) or Kruskal-Wallis (multiple groups) followed by an appropriate post test. A P value of less than 0.05 was deemed to be significant and indicated with either + (for comparisons against vehicle non-LPS-treated groups) or * (for comparisons against vehicle LPS-treated groups).

Effect of compound 2 and dexamethasone on cytokine protein expression from LPS-stimulated THP-1 cells. THP-1 cells were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], compound 2 (0.1 nM to 1 μM), or dexamethasone (1 μM) before stimulation with vehicle [0.01% (v/v) PBS], or LPS (0.1 μg/ml). Twenty-four hours later, the cell culture supernatants were collected and stored at –80°C until required for cytokine assessment by ELISA (A, TNFα; B, IL-8; C, GROα; and D, MIP-1α). Results are expressed as mean ± S.E. mean of three experiments run in duplicate. Statistical significance between groups was assessed using Student's t test (two groups) or Kruskal-Wallis (multiple groups) followed by an appropriate post test. A P value of less than 0.05 was deemed to be significant and indicated with either + (for comparisons against vehicle non-LPS-treated groups) or * (for comparisons against vehicle LPS-treated groups).
Determination of the Effect of SB 239063 and Compound 2 on LPS-Induced Airway Inflammation in the Rat. LPS exposure caused an increase in lung tissue neutrophilia and eosinophilia (Fig. 8) and BALF neutrophil (from 15 ± 5 to 1849 ± 174 × 103 cell/ml) and eosinophil (from 1 ± 0 to 68 ± 12 × 103 cell/ml) number. Oral administration of the two p38 MAPK inhibitors and dexamethasone caused a dose-related reduction of LPS-induced lung tissue neutrophilia and eosinophilia (Fig. 8). The impact of the compounds on tissue neutrophilia was confirmed by measuring MPO levels, which are specific markers of this cell type (Fig. 8). Neither p38 MAPK inhibitor had a significant effect on the number of neutrophils or eosinophils in the airway lumen, with the exception of the highest dose of SB 239063 (30 mg/kg), which significantly reduced neutrophilia and eosinophilia (797 ± 128 and 22 ± 3 × 103 cell/ml, respectively). Dexamethasone treatment (1 mg/kg) significantly reduced levels of LPS-induced recruitment of neutrophils and eosinophils in the lavage fluid (481 ± 68 and 21 ± 6 × 103 cell/ml, respectively). There was no significant effect of any of the compounds on BALF and lung tissue lymphomononuclear cell number (data not shown).

Effect of SB 239063 and dexamethasone on cytokine protein expression from LPS-stimulated human lung tissue macrophages. Human lung tissue macrophages were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], SB 239063 (10 nM to 10 μM), or dexamethasone (1 μM) before stimulation with vehicle [0.01% (v/v) PBS] or LPS (0.1 μg/ml). Twenty-four hours later, the cell culture supernatants were collected and stored at –80°C until required for cytokine assessment by ELISA (A, TNFα; B, IL-8; C, IL-6; D, MIP-1α; E, GM-CSF; and F, G-CSF). Results are expressed as mean ± S.E. mean run in triplicate. Statistical significance between groups was assessed using Student's t test (two groups) or Kruskal-Wallis (multiple groups) followed by an appropriate post test. A P value of less than 0.05 was deemed to be significant and indicated with either + (for comparisons against vehicle non-LPS-treated groups) or * (for comparisons against vehicle LPS-treated groups).

Effect of compound 2 and dexamethasone on cytokine protein expression from LPS-stimulated human lung tissue macrophages. Human lung tissue macrophages were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], compound 2 (10 nM to 10 μM), or dexamethasone (1 μM) before stimulation with vehicle [0.01% (v/v) PBS] or LPS (0.1 μg/ml). Twenty-four hours later, the cell culture supernatants were collected and stored at –80°C until required for cytokine assessment by ELISA (A, TNFα; B, IL-8; C, IL-6; D, MIP-1α; E, GM-CSF; and F, G-CSF). Results are expressed as mean ± S.E. mean run in triplicate. Statistical significance between groups was assessed using Student's t test (two groups) or Kruskal-Wallis (multiple groups) followed by an appropriate post test. A P value of less than 0.05 was deemed to be significant and indicated with either + (for comparisons against vehicle non-LPS-treated groups) or * (for comparisons against vehicle LPS-treated groups).
Assessment of TNFα and IL-1β protein levels showed that the LPS-induced increase in TNFα, but not IL-1β [saline/vehicle, 158 ± 23; LPS/vehicle, 1720 ± 183; LPS/compound 2 (30 mg/kg), 1633 ± 248; LPS/SB 239063 (30 mg/kg), 1434 ± 199; LPS/dexamethasone (1 mg/kg), 532 ± 88 pg/mg lung tissue], was inhibited by both p38 MAPK inhibitors in a dose-related fashion (data obtained with the top dose of compound shown in Fig. 9A), whereas the positive control, dexamethasone, significantly reduced levels of both cytokines. Using p65 DNA binding to assess the level of NF-κB pathway activation, the data showed a marked increase after LPS exposure, which was not affected by either of the p38 MAPK inhibitors but was abolished by treatment with dexamethasone (Fig. 9B). Exposure to LPS caused an increase in TNFα gene expression, which was significantly reduced by dexamethasone but not impacted on by either of the p38 MAPK inhibitors (Fig. 9C). There was a marked increase in levels of phosphorylated Mnk1 in the lung tissue homogenate from animals previously exposed to LPS (see example in Fig. 9D). Treatment with either p38 MAPK inhibitor blocked this increase in phospho-Mnk1 (Fig. 9D).
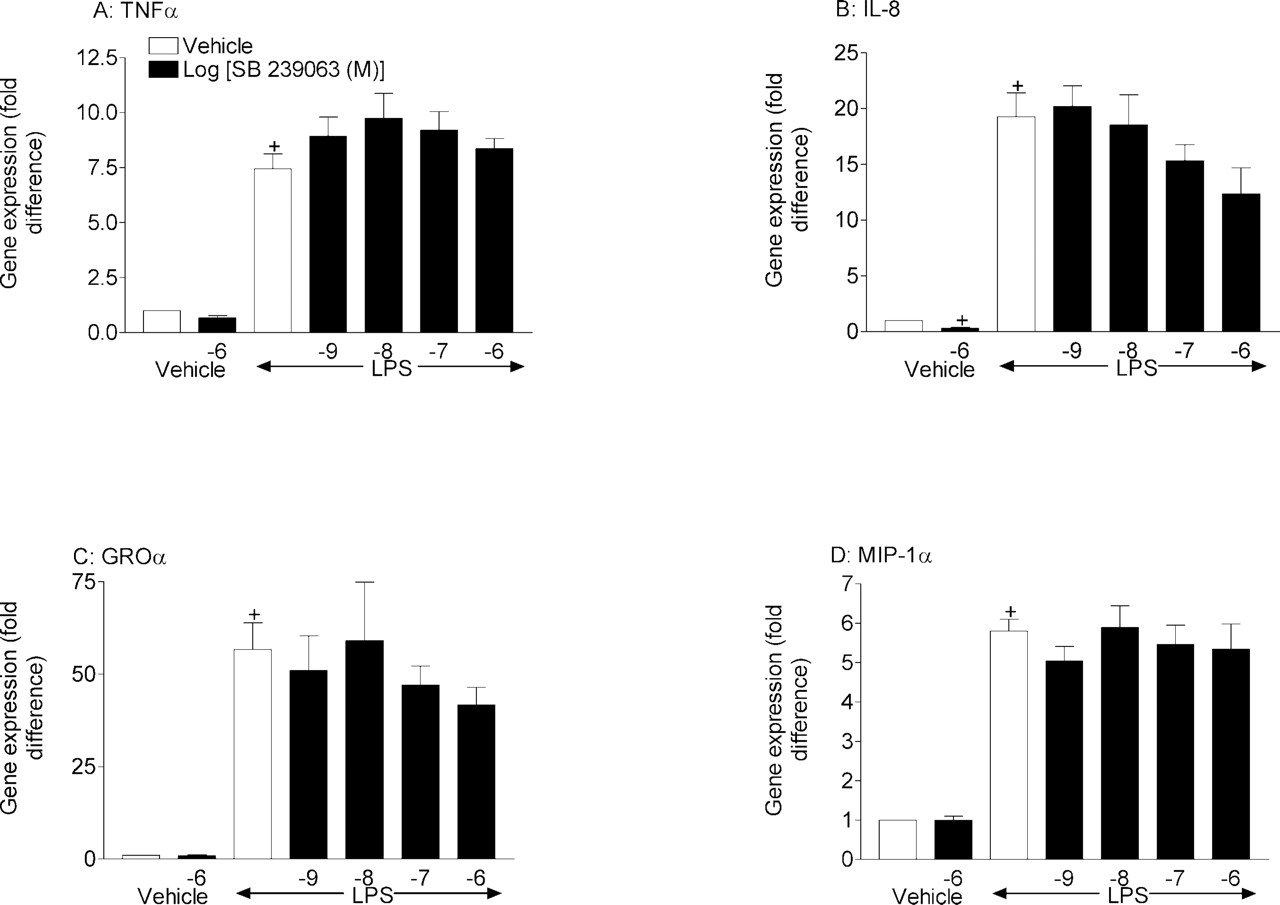
Effect of SB 239063 and dexamethasone on cytokine gene expression from LPS-stimulated THP-1 cells. THP-1 cells were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], SB 239063 (1 nM to 1 μM), or dexamethasone (1 μM) before stimulation with vehicle [0.01% (v/v) PBS] or LPS (0.1 μg/ml). Two hours later, the cell suspension was centrifuged, and cell pellets were stored at –80°C until required for cytokine gene expression by real time PCR (A, TNFα; B, IL-8; C, GROα; and D, MIP-1α). Results are expressed as mean ± S.E. mean three experiments run in duplicate. Statistical significance between groups was assessed using Student's t test (two groups) or Kruskal-Wallis (multiple groups) followed by an appropriate post test. A P value of less than 0.05 was deemed to be significant and indicated with either + (for comparisons against vehicle non-LPS-treated groups) or * (for comparisons against vehicle LPS-treated groups).
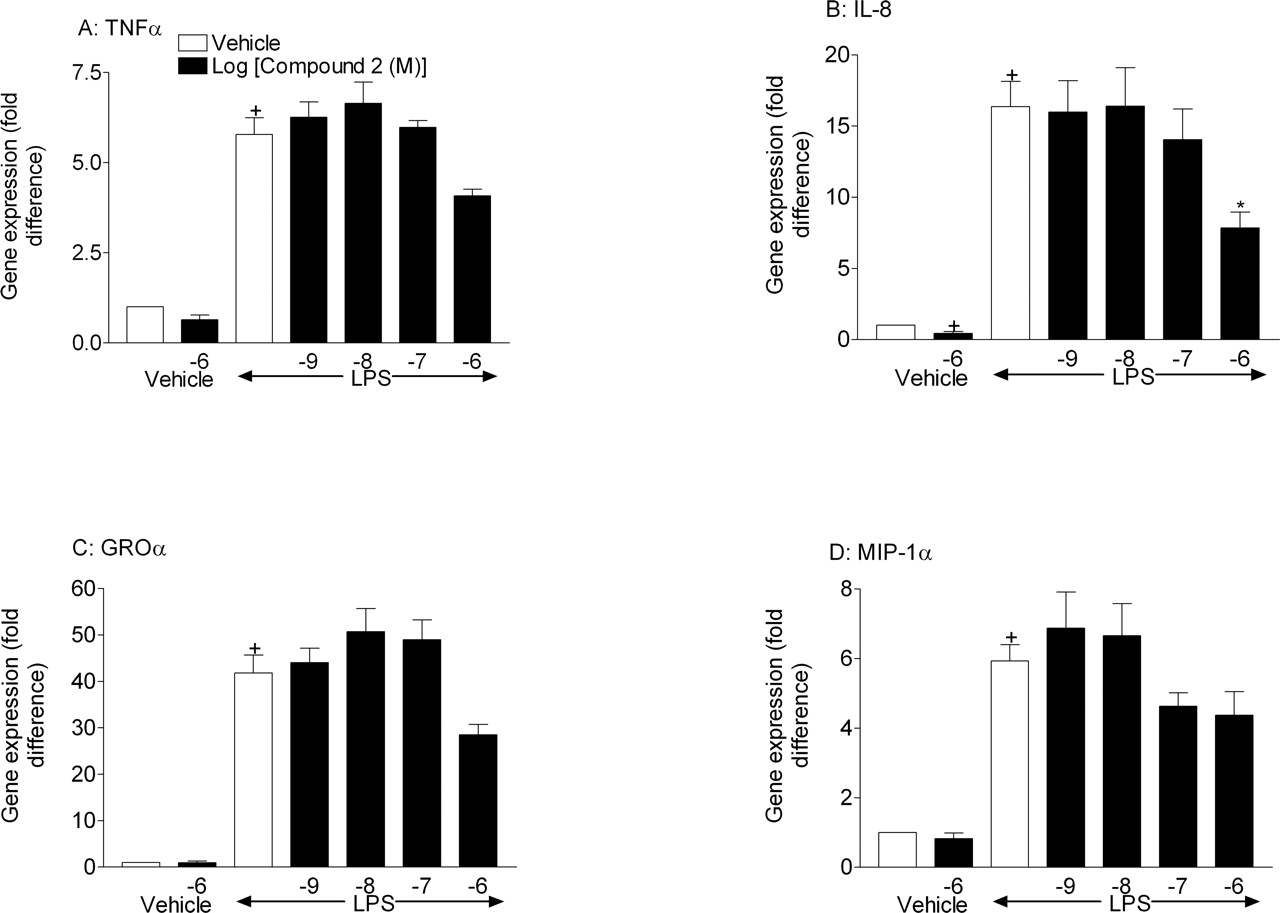
Effect of compound 2 and dexamethasone on cytokine gene expression from LPS-stimulated THP-1 cells. THP-1 cells were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], compound 2 (1 nM to 1 μM), or dexamethasone (1 μM) before stimulation with vehicle [0.01% (v/v) PBS] or LPS (0.1 μg/ml). Two hours later, the cell suspension was centrifuged, and cell pellets were stored at –80°C until required for cytokine gene expression by real-time PCR (A, TNFα; B, IL-8; C, GROα; and D, MIP-1α). Results are expressed as mean ± S.E. mean three experiments run in duplicate. Statistical significance between groups was assessed using Student's t test (two groups) or Kruskal-Wallis (multiple groups) followed by an appropriate post test. A P value of less than 0.05 was deemed to be significant and indicated with either + (for comparisons against vehicle non-LPS-treated groups) or * (for comparisons against vehicle LPS-treated groups).
Discussion
The role of p38 MAPK in the expression of inflammatory cytokines is not clear; a recent review indicates this kinase may regulate their expression transcriptionally, post-transcriptionally, translationally, or post-translationally (Newton and Holden, 2003). Interpretation of some of the published data is further complicated by the use of inhibitors at single, high concentrations. The aim of this study was to determine the impact of two second-generation p38 MAPK inhibitors, SB 239063 (Underwood et al., 2000a,b) and compound 2, on the expression of a range of inflammatory cytokines at the gene and protein levels.

Effect of SB 239063 and compound 2 on the expression of a marker of p38 MAPK activity and protein translation-phosphorylated Mnk1 in LPS-stimulated THP-1 cells. THP-1 cells were pretreated (1 h) with vehicle [0.1% (v/v) DMSO], SB 239063 (1 nM to 1 μM), or compound 2 (1 nM to 1 μM) before stimulation with vehicle [0.01% (v/v) PBS] or LPS (0.1 μg/ml). Two hours later, the cell pellet was collected, the protein extracted, and Western blot analysis performed. A is a representative Western blot from one experimental run, and B is the mean of the results expressed as mean ± S.E. mean of three experiments.

Effect of SB 239063, compound 2, and dexamethasone on LPS-induced cellular accumulation and myeloperoxidase levels in tissue. Rats were treated with vehicle (0.5% methylcellulose and 0.2% Tween 80 in distilled water, 2 ml/kg), SB 239063 (3, 10, or 30 mg/kg), compound 2 (1, 3, 10, or 30 mg/kg), or dexamethasone (0.01, 0.1, or 1 mg/kg) orally 1 h prior to aerosolized saline or LPS (0.3 mg/ml, 30 min). Six hours after challenge, the animals were culled and inflammatory status of the airways assessed. The number of tissue neutrophils (A) and eosinophils (B) was determined, and tissue MPO activity (C) was determined using a colorimetric assay. Results represent mean ± S.E. mean (n = 8). Statistical significance between vehicle groups was assessed using Student's t test with a Mann-Whitney post test (+, P < 0.05). Statistical significance between LPS-treated groups was assessed by one-way ANOVA followed by the appropriate post test. *, P < 0.05 compared with LPS-exposed, vehicle-dosed control group.
In LPS-stimulated human monocytes and lung tissue macrophages, pretreatment with both compounds reduced the protein level of most cytokines measured, a function of inhibiting p38 MAPK observed by other groups in these cell types (Lee et al., 1994; Carter et al., 1999; Shafer et al., 1999; Nick et al., 2000; Underwood et al., 2000a,b; Campbell et al., 2004). It appeared that the efficacy and potency of the compounds tended to be greater on the monocytic cell line compared with the primary lung tissue macrophages. This difference was particularly evident for IL-8 protein expression, a result that is in agreement with a recent study showing only limited inhibition of IL-8 production by SB 203580 from alveolar macrophages (Koch et al., 2003). The reason for this difference it not clear, but it could mean a less dominant role for p38 MAPK in this cell type, as suggested by Carter et al. (1999) and Nick et al. (2000), and is something to consider in the development of compounds of this type for the treatment of diseases such as chronic obstructive pulmonary disease, where macrophages may be an important effector cell.
TaqMan real-time mRNA expression analysis, which is more sensitive than traditional PCR, demonstrated that the compounds had limited effect on the mRNA levels of the same cytokines. Where effects on gene expression were observed, it was evident only at higher concentrations than necessary to impact on protein expression. Interestingly, both compounds appeared to inhibit basal and LPS-stimulated IL-8 gene expression, which may suggest a different role for p38 MAPK for this cytokine. Overall, this data would concur with results published by Young et al. (1993), Lee et al. (1994), and Kotlyarov et al. (1999), inasmuch as the predominant effect of p38 inhibitors on cytokine protein production is via a blockade of post-transcriptional mechanisms. Indeed, the data from the in vitro and in vivo studies demonstrate an inhibition of phosphorylation of Mnk1, which indicates first that the two compounds are inhibiting p38 MAPK in the two systems, and second, that the impact of these inhibitors is at the level of protein translation. The data in the human cell-based assay demonstrate clearly that the suppression of cytokine production is paralleled by the inhibition of a marker of protein translation (Pyronnet et al., 1999; Waskiewicz et al., 1999), phosphorylation of Mnk1, which suggests a causative association. In the tissue from the preclinical model, similar inhibition of Mnk1, and not NF-κB pathway activity or cytokine gene expression, was observed, which again would suggest the impact of these compounds is at the level of protein translation.

Effect of SB 239063, compound 2, and dexamethasone on TNFα cytokine protein levels in the BAL fluid, p65 DNA binding, TNFα cytokine gene levels, and phosphorylated Mnk1 levels in the lung tissue. Rats were treated with vehicle (0.5% methylcellulose and 0.2% Tween 80 in distilled water, 2 ml/kg), SB 239063 (3, 10, or 30 mg/kg), compound 2 (1, 3, 10, or 30 mg/kg), or dexamethasone (0.01, 0.1, or 1 mg/kg) orally 1 h prior to aerosolized saline or LPS (0.3 mg/ml, 30 min). Six hours after challenge, the animals were culled and protein levels were determined from the BAL fluid by ELISA for TNFα (A). Lung p65 DNA binding was determined 6 h after challenge by an ELISA-based assay (B), and TNFα gene expression was measured using TaqMan real-time PCR (C). Western blot analysis was performed on lung tissue homogenates using antibodies raised against phosphorylated Mnk1 (D is a scan of a representative blot). Results represent mean ± S.E. mean (n = 8). Statistical significance between vehicle groups was assessed using students t test with a Mann-Whitney post test (+, P < 0.05). Statistical significance between LPS-treated groups was assessed by one-way ANOVA followed by the appropriate post test. *, P < 0.05 compared with LPS-exposed, vehicle-dosed control group.
Other studies outlined in the Introduction have suggested an impact of p38 MAPK inhibitors on cytokine gene transcription, and indeed, at higher compound concentrations, similar data have been observed in this study. It would seem from this study that inhibition of the “transcriptional aspects” of cytokine expression using p38 MAPK inhibitors is only achieved at high concentrations, which may question the pharmacological/therapeutic relevance of this feature of this class of anti-inflammatory compound.
In the preclinical rodent model of LPS-driven airway inflammation, both compounds inhibited the recruitment of innate effector cells, confirming similar results from studies by our group and others with p38 MAPK inhibitors (Nick et al., 2000, 2002; Underwood et al., 2000a,b; Haddad et al., 2001). Both compounds inhibited LPS-induced TNFα protein production in the lung, as shown by Nick et al. (2000), but interestingly, unlike in our previous study with SB 203580, they had no effect on IL-1β protein levels (Haddad et al., 2001). This result would suggest that using this model protocol, the LPS-induced production of TNFα and not IL-1β involves p38 MAPK. The reason for the difference between our previous study and this one is not known but may be due to the lack of selectivity of SB 203580 (Dean et al., 1999; Lali et al., 2000). In an attempt to determine whether the compounds impacted transcriptionally or translationally in this model, we assessed the impact of the compounds on the NF-κB pathway by measuring p65 DNA binding by an ELISA-based assay and TNFα gene expression by TaqMan real-time PCR and Mnk1 phosphorylation by Western blot analysis. The data clearly show that, whereas the positive control dexamethasone significantly inhibits both p65 DNA binding and TNFα gene expression, neither compound appears to impact at the level of transcription. This result demonstrates that p38 MAPK regulates cytokine protein production in the lungs, after exposure to aerosolized LPS, through post-transcriptional mechanisms.
In summary, we have determined the impact of two second-generation p38 MAPK inhibitors in LPS-driven human cell-based assays and in a preclinical rodent model of endotoxin induced innate response in the airway. The data shown here demonstrate that, although at high compound concentrations there is some level of transcriptional regulation, the predominant role of p38 MAPK in cytokine production is at the translational level.
Footnotes
- Received July 27, 2005.
- Accepted December 15, 2005.
-
doi:10.1124/jpet.105.093310.
-
ABBREVIATIONS: MAPK, mitogen-activated protein kinase; SB 203580, 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole; LPS, lipopolysaccharide; TNFα, tumor necrosis factor α; IL, interleukin; NF-κB, nuclear factor κB; ROCK, Rho-associated coiled-coil forming protein serine/threonine kinase; Alk5, activin receptor-like kinase; AKT, serine/threonine kinase (protein kinase B); Lck, light chain kinase; ERK, extracellular signal-regulated protein kinase; MEK, MAPK/ERK kinase; SB 239063, trans-1-(4-hydroxycyclohexyl)-4-(4-fluorphenyl)-5-(2-methoxy-pyridimidin-4-yl)imidazole; compound 2, 1-(1,3-dihydroxyprop-2-yl)-4-(4-fluorophenyl)-5-(2-phenoxypyrimidin-4-yl)imidazole); PBS, phosphate-buffered saline; ELISA, enzyme-linked immunosorbent assay; MIP, macrophage inflammatory protein; GRO, growth-regulated oncogene product; GM-CSF, granulocyte macrophage–colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor; PCR, polymerase chain reaction; MOPS, 4-morpholinepropanesulfonic acid; MPO, myeloperoxidase; BALF, bronchoalveolar lavage fluid; BAL, bronchoalveolar lavage.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}