





Abstract
Activin-A is a pleiotropic cytokine belonging to the transforming growth factor-β superfamily and has been implicated in asthma and pulmonary fibrosis. However, the role of activin-A and its endogenous inhibitor, follistatin, in the pathogenesis of chronic obstructive pulmonary disease (COPD) is unknown.
We first quantified activin-A and follistatin in the lungs of air- or cigarette smoke-exposed mice and in the lungs of patients with COPD by immunohistochemistry, ELISA and quantitative real-time PCR. We subsequently studied the effect of cigarette smoke on primary human bronchial epithelial cells in vitro. Next, activin-A signalling was antagonised in vivo by administration of follistatin in mice exposed to air or cigarette smoke for 4 weeks.
Protein levels of activin-A were increased in the airway epithelium of patients with COPD compared with never-smokers and smokers. Cigarette smoke-exposed human bronchial epithelial cells expressed higher levels of activin-A and lower levels of follistatin. Both mRNA and protein levels of activin-A were increased in the lungs of cigarette smoke-exposed mice, whereas follistatin levels were reduced upon cigarette smoke exposure. Importantly, administration of follistatin attenuated the cigarette smoke-induced increase of inflammatory cells and mediators in the bronchoalveolar lavage fluid in mice.
These results suggest that an imbalance between activin-A and follistatin contributes to the pathogenesis of cigarette smoke-induced inflammation and COPD.
Abstract
Imbalance of activin-A and FST in favour of activin-A signalling in COPD patients cigarette smoke-exposed mice http://ow.ly/qMqVN
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory disease with obstruction of the small airways (obstructive bronchiolitis) and destruction of lung parenchyma (emphysema). The major risk factor for COPD is tobacco smoking, but air pollution and occupational exposure to dusts and chemicals are also important contributors, together with genetic susceptibility [1]. COPD is a leading cause of morbidity and mortality worldwide [2]. Besides smoking cessation, current treatment of COPD is mainly symptomatic. It is believed that a complex interaction between a dysregulated immune response, oxidative stress, imbalanced proteolytic activity and increased apoptosis leads to the pathology seen in patients with COPD [3–5]. To date, the precise molecular mechanisms underlying the pathogenesis of COPD are not fully understood.
Members of the transforming growth factor (TGF)-β superfamily, which includes TGF-β, activins and bone morphogenetic proteins (BMPs), are pleiotropic cytokines regulating fundamental physiological processes and have been linked to numerous diseases [6]. Since TGF-β has a key role in immune regulation and tissue remodelling in COPD, other members of the TGF-β superfamily may also be involved in the pathogenesis of COPD [7–9]. Activin-A is a homodimeric protein comprising of two inhibin βA subunits (INHBA). It signals via a constitutively active activin type II receptor (Act-RIIA and Act-RIIB) that recruits and phosphorylates an activin type I receptor (activin receptor-like kinases-4 and/or -7). The receptor complex propagates the signal by phosphorylating the same intracellular Smads (Smad2 and Smad3) as TGF-β. Phosphorylated Smad2/3 subsequently translocate to the nucleus in association with Smad4 to initiate gene transcription [10, 11]. Regulation of activin-A signalling is achieved by the endogenous inhibitor follistatin, which binds activin-A with high affinity and blocks the interaction between activin-A and the type II receptor [12]. Follistatin can also bind and inhibit several BMPs (BMP-2, -4, -5, -6, -7 and -8) and TGF-β3, although the binding affinity of follistatin is much higher for activin-A than for BMPs [13, 14].
Several groups have demonstrated activation of the activin-A signalling pathway in an experimental model of allergic asthma and in patients with asthma [15–18]. Similarly, patients with acute respiratory distress syndrome or pulmonary fibrosis have higher levels of activin-A in bronchoalveolar lavage (BAL) fluid and lung tissue, respectively, compared to control patients [19, 20].
Since activin-A is an important inflammatory mediator in several respiratory diseases, we hypothesised that activin-A plays a significant role in the pathogenesis of COPD. To elucidate this, we investigated the expression of activin-A and follistatin in lungs of never-smokers and smokers with and without COPD, and in primary human bronchial epithelial cell (HBEC) cultures exposed to cigarette smoke. Finally, we used a cigarette smoke-induced mouse model of COPD to examine the in vivo functional role of activin-A by administering its endogenous inhibitor follistatin.
Materials and methods
A detailed description of the materials and methods can be found in the online supplementary material.
Human study populations
Lung resection specimens were obtained from 100 patients, of which 89 were from surgery for solitary pulmonary tumours (Ghent University Hospital, Ghent, Belgium) and 11 were from explant lungs of end-stage COPD patients undergoing lung transplantation (University Hospital Gasthuisberg, Leuven, Belgium). Lung tissue of 64 (out of 100) patients, including 11 never-smokers, 24 smokers without airflow limitation and 29 patients with COPD Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage II, was used for mRNA expression analysis (table 1). Lung tissue of 71 (out of 100) patients, including 10 never-smokers, 26 smokers without airflow limitation and 35 patients with COPD (21 GOLD stage II, four GOLD stage III and 10 GOLD stage IV), was used for immunohistochemical analysis (table 2). Lung tissue of 35 (out of 100) patients was used for both mRNA and immunohistochemical analysis (fig. E1).
Written informed consent was obtained from all subjects. This study was approved by the medical ethical committees of Ghent University Hospital and University Hospital Gasthuisberg Leuven.
Activin-A immunohistochemistry and quantification
Paraffin-embedded sections of human lung tissue were subjected to activin-A staining using anti-activin-A antibody as previously described [21]. Immunohistochemical staining with anti-activin-A (R&D Systems, Abingdon, UK) was performed on paraffin sections of the left lung of mice. The area with positive activin-A staining was quantified using KS400 software (Zeiss, Oberkochen, Germany) and was normalised to the length of the basement membrane.
Phospho-Smad2 immunohistochemistry
Paraffin sections were incubated overnight with primary phospho-Smad2 (Ser465/467) antibodies (Cell Signaling Technology, Danvers, MA, USA) or isotype rabbit IgG (Abcam, Cambridge, UK). Next, the slides were incubated with PowerVision poly-horseradish peroxidase-anti-rabbit (Immunovision Technologies, Burlingame, CA, USA) and stained with 3,3'-diaminobenzidine substrate (Dako, Glostrup, Denmark). Finally, sections were rinsed in demineralised water, counterstained with Mayer's haematoxylin (Sigma-Aldrich, St. Louis, MO, USA) and mounted in DPX (Klinipath, Duiven, the Netherlands).
Culture of human bronchial epithelial cells
Primary HBECs were obtained by enzymatic digestion from lung resection specimens obtained from anonymous donors during surgery for lung cancer, as described previously [22]. Cells from passage 2 were cultured at an air–liquid interface in medium containing a high concentration of retinoic acid to induce mucociliary differentiation, as described previously [23].
After the 14-day air–liquid interface culture period, HBECs were exposed to whole volatile smoke or air as a negative control, using a modification of the system reported by Beisswenger et al. [24]. In other experiments, HBECs were stimulated both on the apical and basal side with recombinant human activin-A (R&D Systems) or with recombinant human TGF-β1 (R&D Systems) as a positive control.
Cigarette smoke exposure
C57BL/6 mice (Charles River Laboratories, Brussels, Belgium) were exposed to cigarette smoke, as described previously [25].
Administration of follistatin
C57BL/6 mice were injected intraperitoneally with 1 μg of recombinant mouse follistatin-288 (R&D Systems) or PBS (controls) [26]. Mice were injected three times a week in a subacute (4 weeks) cigarette smoke experiment, 30 min before air or cigarette smoke exposure.
Quantitative RT-PCR, ELISA, CBA and flow cytometry
Detailed information can be found in the online supplementary material.
Statistical analysis
Statistical analysis was performed with Sigma Stat software (SPSS 19.0, Chicago, IL, USA), using the Kruskal–Wallis, Mann–Whitney U-test, Wilcoxon Signed Rank, Fisher's exact test and Spearman correlation analysis. Characteristics of the study population are expressed as median (interquartile range). Pearson correlation and linear regression analysis were performed on log-transformed data. Values from the in vitro study with HBECs are reported relative to the control group. Reported values are expressed as mean±sem. A p-value <0.05 was considered to be statistically significant.
Results
Activin-A and follistatin mRNA expression in lungs of patients with COPD
To characterise the expression of activin-A and follistatin in human lung tissue we extracted mRNA from total lung tissue of a study population containing 11 never-smokers, 24 smokers without airflow limitation and 29 patients with COPD GOLD stage II. The characteristics of the different study groups are summarised in table 1. Activin-A is a homodimer of inhibin βA subunits. By measuring the mRNA expression of INHBA, we observed higher mRNA levels of activin-A in lung tissue of current smokers both with and without COPD, compared to never-smokers. Moreover, current smokers had significantly higher activin-A mRNA expression, compared to ex-smokers (fig. 1a). The mRNA expression of follistatin (fig. 1b) was similar between the different study groups. Taken together, the ratio of activin-A to follistatin is increased in current smokers both with and without COPD in comparison with never-smokers (fig. 1c).

Pulmonary mRNA expression of INHBA (inhibin βA subunit of activin-A) and follistatin (FST) in human subjects. mRNA levels of a) INHBA and b) FST in the total lung of 11 never-smokers, 12 current smokers,12 ex-smokers, 17 current smokers with chronic obstructive pulmonary disease (COPD) and 12 ex-smokers with COPD, as measured by quantitative real-time PCR. c) The ratio of INHBA/FST mRNA expression. mRNA levels were corrected using a calculated normalisation factor based on mRNA expression of three reference genes (GAPDH, PPIA and HPRT-1). Data are presented as box and whisker plots. *: p<0.05; **: p<0.01; #: p=0.053.
Increased activin-A signalling in airway epithelium of patients with COPD
In order to localise pulmonary activin-A signalling, we performed immunohistochemical staining for activin-A and phospho-Smad2 on lung tissue sections of never-smokers, smokers and patients with COPD GOLD stage II–IV. The demographic, clinical and lung functional characteristics of the study subjects are presented in table 2. Figure 2 shows representative lung sections stained for activin-A in never-smokers (fig. 2a), smokers (fig. 2b), patients with COPD GOLD stage II (fig. 2C) and patients with COPD GOLD stage IV (fig. 2d and f). We observed positive staining for activin-A in bronchiolar epithelial cells with a minor staining of smooth muscle cells and alveolar macrophages in the lungs of never-smokers (fig. 2a). Using imaging analysis software, we quantified the activin-A positive staining in the airway epithelium. Protein levels of activin-A in the airway epithelium were significantly increased in patients with COPD GOLD stage II and III–IV compared to never-smokers and smokers without airflow limitation (fig. 2m). The expression of activin-A in the airway epithelium is significantly associated with disease severity, expressed as post-bronchodilator forced expiratory volume in 1 s (FEV1) % predicted and FEV1/forced vital capacity % (table E1, fig. E2). After adjustment for various confounders, the association with FEV1 remained significant (table E2). After semi-quantitative scoring, we also demonstrated higher scores for activin-A in the airway smooth muscle cell layer and alveolar macrophages of patients with COPD compared to never-smokers (fig. E3).

Pulmonary a–f) activin-A and g–l) phospho-Smad2 expression in human lungs by immunohistochemistry. Representative photomicrographs for human lung sections of: a and g) a never-smoker; b and h) a smoker without airflow limitation; c and i) a patient with chronic obstructive pulmonary disease (COPD) Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage II; and d and j) a patient with COPD GOLD stage IV. e and k) An isotype control. f and l) Enlargement of the boxed area in d) and j), respectively. Scale bars=100 μm. m) Quantification of activin-A protein expression in airway epithelium of 10 never-smokers, 26 smokers, 21 patients with COPD GOLD stage II and 14 patients with COPD GOLD stage III–IV. Results are expressed as the epithelial area positive for activin-A normalised to the length of the basement membrane (Pbm). Data are presented as box and whisker plots. *: p<0.05; **: p<0.01.
We evaluated the phosphorylation status and nuclear accumulation of Smad2 as a hallmark for activated activin-A signalling. Almost no positive nuclear staining was present in the airway epithelial cells of never-smokers and smokers without COPD (fig. 2g and h), whereas, nuclei of airway epithelial cells of patients with COPD stained strongly (fig. 2i–l). Importantly, there was a significant, positive correlation between the semi-quantitative levels of p-Smad2 and activin-A in the airway epithelium (fig. E4).
Cigarette smoke exposure increases expression of activin-A in HBECs
To confirm the results seen in patients with COPD, primary HBECs, grown in vitro at air–liquid interface, were exposed to mainstream cigarette smoke. HBECs and basal media were harvested 3, 6 and 24 h after cigarette smoke exposure. RT-PCR analysis on mRNA extracted from HBECs revealed a significant increase in expression of activin-A 3, 6 and 24 h after cigarette smoke exposure, compared to air-exposed cells (fig. 3a). The follistatin mRNA expression was slightly lower in cigarette smoke-exposed HBECs (fig. 3b). This resulted in a significantly increased ratio of activin-A to follistatin in cigarette smoke-exposed HBECs compared to air-exposed HBECs (fig. 3c). Accordingly, the release of activin-A in the basal medium from cigarette smoke-exposed HBEC cultures was augmented, especially 24 h after cigarette smoke exposure (fig. 3d), whereas the release of follistatin was decreased after cigarette smoke exposure (fig. 3e). In conclusion, the ratio of activin-A to follistatin was significantly increased in the basal medium of cigarette smoke-exposed HBECs 24 h after cigarette smoke exposure (fig. 3f). These data show that expression and release of activin-A in response to cigarette smoke is enhanced in human HBECs.

Expression of activin-A and follistatin (FST) in air–liquid interface human bronchial epithelial cell (HBEC) cultures upon exposure to air or cigarette smoke. Basal media and cells were harvested 3, 6 and 24 h after exposure. mRNA from HBECs was isolated and analysed for the expression of a) INHBA (inhibin βA subunit of activin-A) and b) FST by RT-PCR. c) The ratio of INHBA to FST mRNA expression. mRNA levels were corrected using a calculated normalisation factor based on mRNA expression of two reference genes (RPL13A and ATP5B). Protein levels of d) activin-A and e) follistatin in the basal medium were analysed by ELISA. f) The ratio of activin-A to follistatin protein. Results are reported relative to the air-exposed group. Data are presented as mean±sem of five different experiments, each performed in duplicate and using cells from different donors. *: p<0.05.
Increased pulmonary expression of activin-A upon cigarette smoke exposure in mice
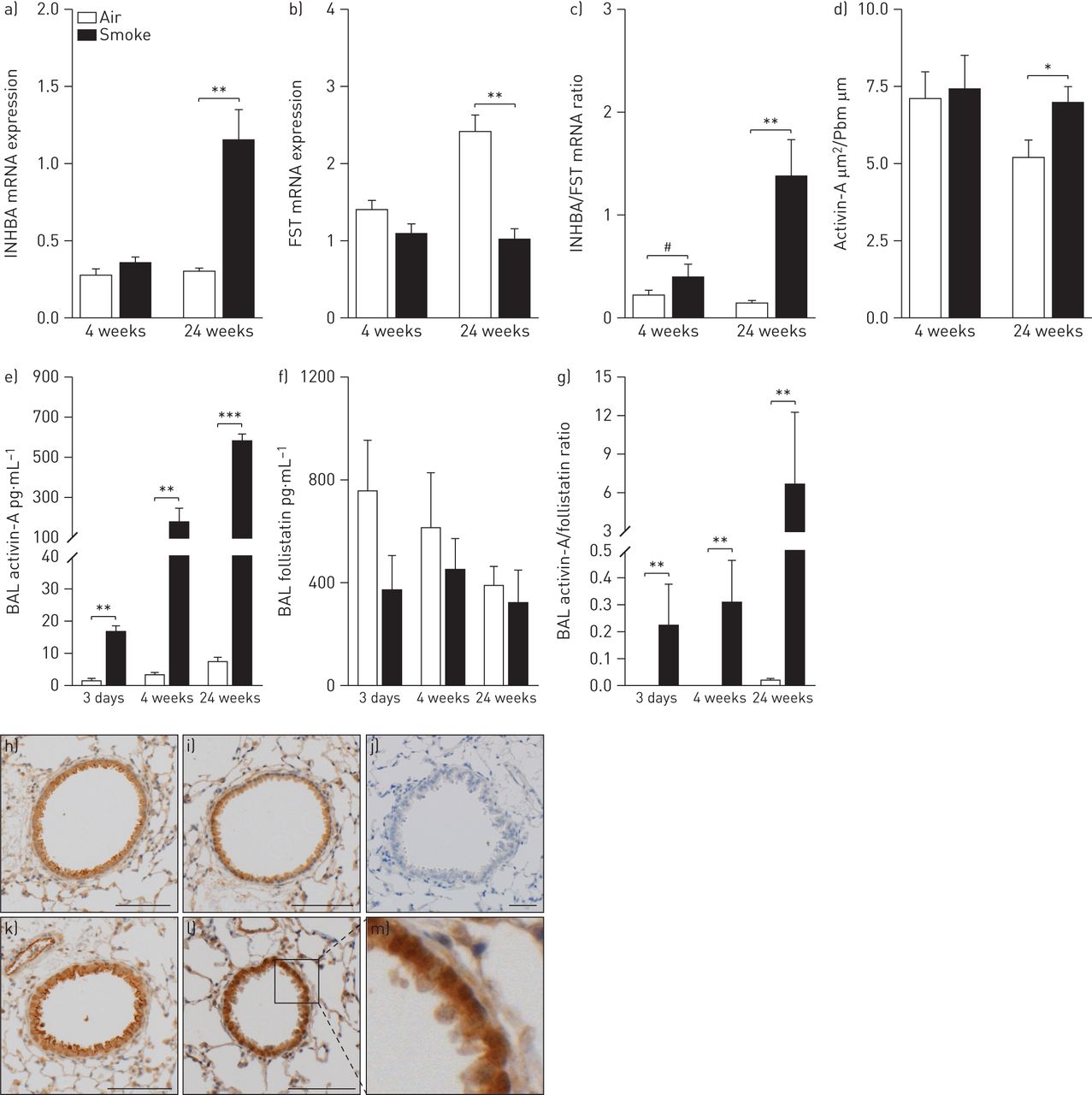
As previously shown by our group [25], exposure of C57BL/6 mice to cigarette smoke for 4 weeks or 24 weeks led to inflammation in the BAL compartment and lung tissue, characterised by increased numbers of macrophages, neutrophils, dendritic cells and B- and T-lymphocytes (figs E6 and E7). The effect of cigarette smoke on the pulmonary expression of activin-A and follistatin was determined in total lung tissue by quantitative RT-PCR and immunohistochemistry and in BAL fluid supernatant by ELISA. Activin-A mRNA expression in lung tissue was significantly increased upon chronic cigarette smoke exposure, compared to air-exposed littermates (fig. 4a), while cigarette smoke significantly downregulated follistatin mRNA expression (fig. 4b). Importantly, immunohistochemistry on lung tissue revealed activin-A staining in airway epithelium (fig. 4h–m), which was increased upon chronic (24 weeks) cigarette smoke exposure (fig. 4d), similar to our observations in patients with COPD. Activin-A protein levels were increased significantly in BAL fluid at all time-points, with an especially marked increase after 24 weeks of cigarette smoke exposure (fig. 4e). In contrast, the protein levels of follistatin in BAL fluid were similar between air- and cigarette smoke-exposed mice (fig. 4f). Taken together, the ratio of activin-A to follistatin was increased after cigarette smoke exposure at all time-points, both on mRNA and protein level (fig. 4c and g).

Expression of activin-A and follistatin (FST) in C57BL/6 mice after acute (3 days), subacute (4 weeks) and chronic (24 weeks) exposure to air or cigarette smoke. mRNA levels a) of INHBA (inhibin βA subunit of activin-A) and b) FST in total lung as measured by quantitative real-time PCR. c) The ratio of INHBA to FST mRNA expression. mRNA levels were corrected using a calculated normalisation factor based on mRNA expression of three reference genes (GAPDH, TFRC and HPRT-1). d) Quantification of activin-A protein expression in the airway epithelium of mice after exposure to air or cigarette smoke. Results are expressed as the epithelial area positive for activin-A normalised to the length of the basement membrane (Pbm). Protein levels of e) activin-A and f) follistatin in bronchoalveolar lavage (BAL) fluid measured by ELISA. g) The ratio of activin-A to follistatin protein. Data are presented as mean±sem (n=8–10 animals per group). *: p<0.05; **: p<0.01; ***: p<0.001; #: p=0.074. h–m) Representative images of activin-A-stained lung sections. h and i) Air-exposed mice at 4 and 24 weeks, respectively. k and l) Cigarette-exposed mice at 4 and 24 weeks, respectively. j) An isotype control. m) Enlargement of the boxed area in l). Scale bars=100 μm.
To determine if activin-A levels persist after smoking cessation, we analysed protein levels of activin-A and numbers of inflammatory cells in BAL fluid of C57BL/6 mice on days 1, 14 and 56 following subacute exposure to air or cigarette smoke (fig. 5). Cigarette smoke-induced inflammation (monocytes, dendritic cells and CD4+ T-lymphocytes) was mostly resolved on day 14 post-exposure, except for neutrophils and CD8+ T-lymphocytes (fig. 5b–d). Inflammation in the BAL fluid was completely cleared 56 days after exposure (fig. 5). Consistent with the inflammation data, there was a notable persistence of activin-A in the BAL fluid on day 14, whereas by day 56 activin-A levels were restored to normal (fig. 5a).

Pulmonary expression of activin-A and pulmonary inflammation in C57BL/6 mice on days 1, 14 and 56 after subacute exposure to air or cigarette smoke. a) Protein levels of activin-A in bronchoalveolar lavage (BAL) fluid measured by ELISA. b–d) Cell differentiation in BAL fluid. b) Neutrophils and c) lymphocytes were counted on cytospins and d) CD8+ T-lymphocytes were enumerated by flow cytometry. Data are presented as mean±sem (n=8 animals per group). *: p<0.05; **: p<0.01.
Cigarette smoke-induced inflammation in BAL is attenuated upon neutralisation of activin-A by follistatin
To elucidate whether activin-A is involved in the pulmonary inflammation upon cigarette smoke exposure, we exposed C57BL/6 mice to cigarette smoke for 4 weeks (subacute exposure) and injected the mice intraperitoneally three times a week with 1 μg follistatin or PBS. In PBS-treated mice, exposure to cigarette smoke significantly increased the total number of BAL cells and absolute numbers of monocytes, neutrophils and CD4+ and CD8+ T-lymphocytes in BAL fluid (fig. 6). In contrast, follistatin treatment significantly attenuated the accumulation of monocytes and CD8+ T-lymphocytes in cigarette smoke-exposed mice (fig. 6b, d and f).
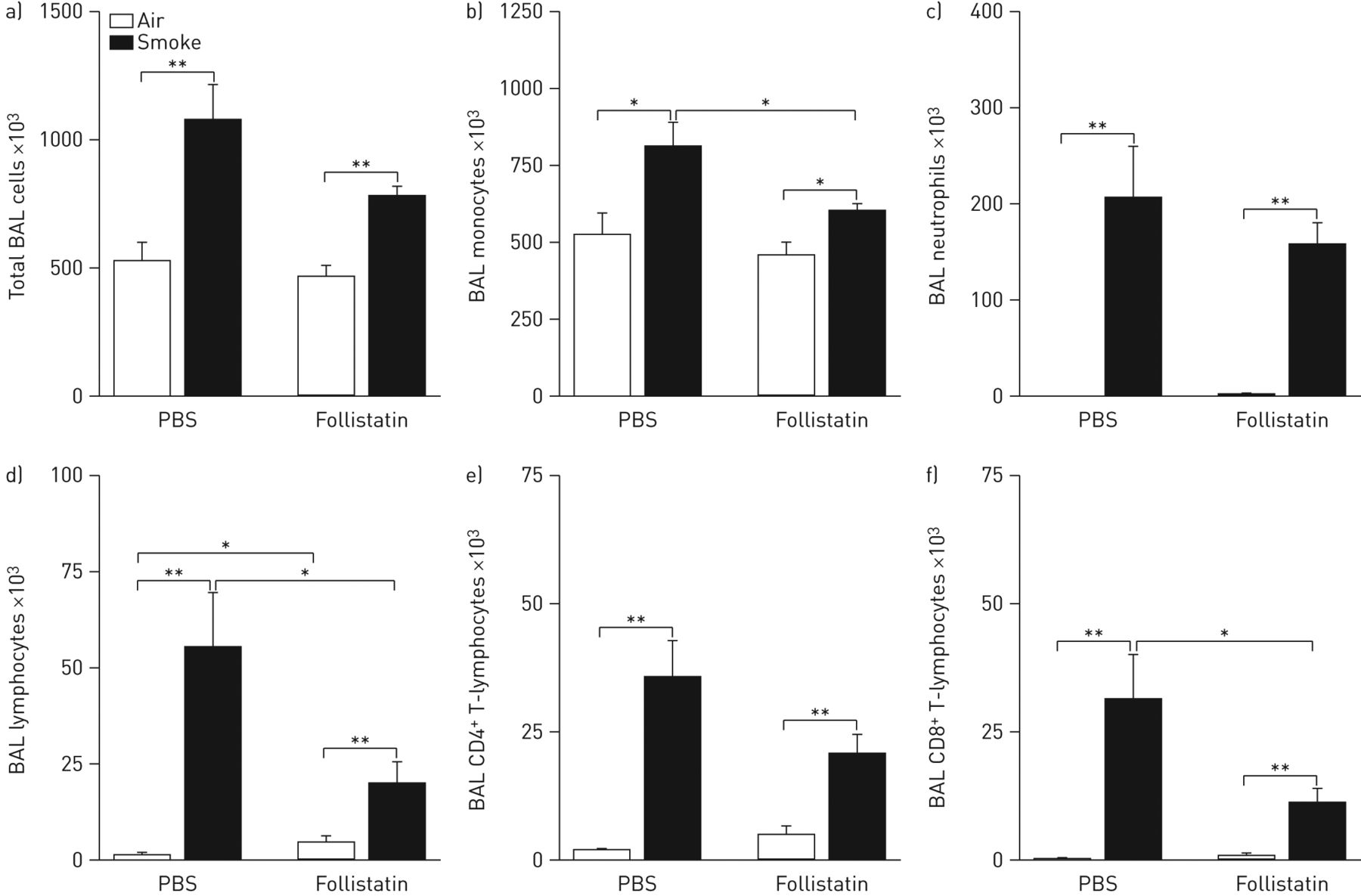
Effect of cigarette smoke exposure and neutralisation of activin-A with follistatin on the total number of bronchoalveolar lavage (BAL) cells and cell subsets in BAL fluid. a) Total BAL cells, b) monocytes, c) neutrophils, d) lymphocytes, e) CD4+ T-lymphocytes and f) CD8+ T-lymphocytes in C57BL/6 mice after 4 weeks of exposure to air or cigarette smoke, by intraperitoneal injection with follistatin or PBS three times a week. All cell types were enumerated by cytospin counts, except for the CD4+ and CD8+ T-lymphocytes which were determined by flow cytometry. Data are presented as mean±sem (n=8 animals per group). *: p<0.05; **: p<0.01.
Follistatin affects cytokine and chemokine expression upon cigarette smoke exposure
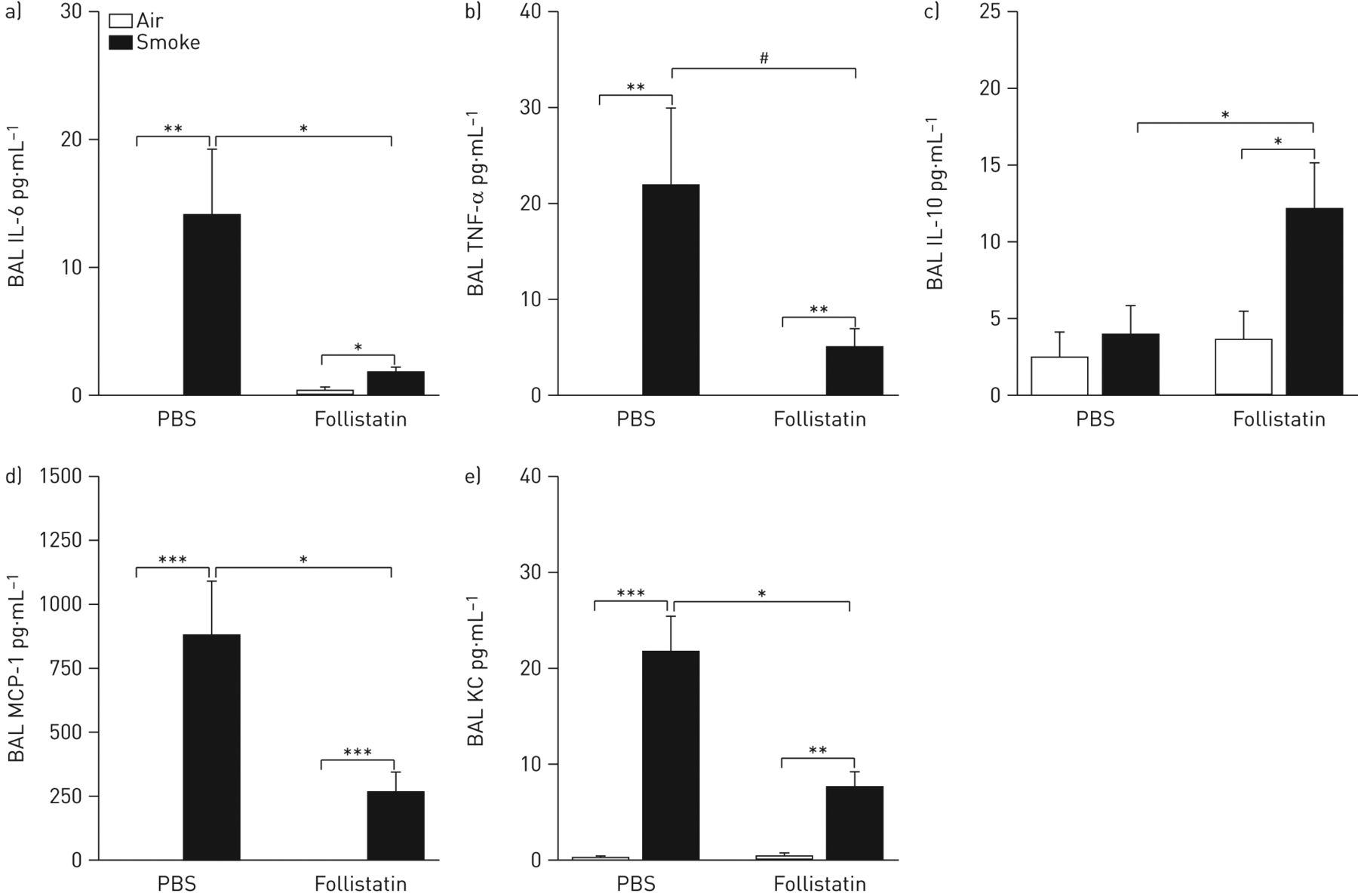
Because we observed a decreased inflammatory cell recruitment in BAL of follistatin-treated mice upon 4 weeks of cigarette smoke exposure, we measured cytokines (interleukin (IL)-6, tumour necrosis factor (TNF)-α and IL-10) and chemokines (monocyte chemotactic protein (MCP)-1 and keratinocyte chemoattractant (KC)) in BAL fluid by cytometric bead array. In PBS-treated mice, cigarette smoke exposure significantly increased the protein levels of IL-6, TNF-α, MCP-1 and KC. In follistatin-treated mice, the increase of IL-6, MCP-1 and KC was significantly attenuated compared to PBS-treated mice (fig. 7a, d and e). Follistatin-treated mice showed a trend towards an attenuation of the cigarette smoke-induced increase of TNF-α in BAL fluid (p=0.061) (fig. 7b). In contrast, IL-10 protein levels did not increase upon cigarette smoke exposure in PBS-treated mice, while follistatin treatment significantly increased IL-10 protein expression upon cigarette smoke exposure (fig. 7c).

Inflammatory cytokines and chemokines in bronchoalveolar lavage (BAL) fluid of C57BL/6 mice after 4 weeks of exposure to air or cigarette smoke, by intraperitoneal injection with follistatin or PBS three times a week. a) Interleukin (IL)-6, b) tumour necrosis factor (TNF)-α, c) IL-10, d) monocyte chemotactic protein (MCP)-1 and e) keratinocyte chemoattractant (KC) protein levels, measured by cytometric bead array. Data are presented as mean±sem (n=8 animals per group). *: p<0.05; **: p<0.01; ***: p<0.001; #: p=0.061.
Follistatin reduces cigarette smoke-induced pulmonary activin-A/TGF-β signalling in mice
We determined activin-A and TGF-β1 protein levels in BAL fluid of PBS- and follistatin-treated mice by ELISA. In follistatin-treated mice, the cigarette smoke-induced increase of activin-A and TGF-β1 was significantly attenuated compared to PBS-treated mice (fig. 8a and b). In contrast, the mRNA expression of activin-A was not influenced by the follistatin treatment (fig. 8d). Using ELISA, we also quantified the phosphorylation state of Smad2 as a marker of active signalling. Cigarette smoke exposure induced a significant increase of p-Smad2 in the lung homogenates of PBS-treated mice, which was strongly suppressed in follistatin-treated mice (fig. 8c). These results indicate that follistatin was effective as an antagonist for activin-A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Activin-A and transforming growth factor (TGF)-β1 signalling in bronchoalveolar lavage (BAL) fluid and the lungs of C57BL/6 mice injected with follistatin or PBS after 4 weeks of exposure to air or cigarette smoke. a) Activin-A, b) TGF-β1 and c) phosphorylated Smad2 (expressed as raw optical density values at 450 nm) levels were quantified by ELISA. d) INHBA (inhibin βA subunit of activin-A) mRNA levels were measured using quantitative real-time PCR. Data are presented as mean±sem (n=8 animals per group). *: p<0.05; **: p<0.01. e) A schematic overview of the effect of cigarette smoke exposure on pulmonary activin-A signalling. Cigarette smoke induces activin-A expression in airway epithelial cells, but attenuates its endogenous antagonist follistatin. Activin-A binds to activin type II receptors on the surface of target cells, which oligomerise with activin type I receptors. The activated type I receptor phosphorylates intracellular protein Smad2, which forms a complex with Smad4 and translocates to the nucleus to initiate gene expression of target genes, such as TGF-β1, which signal through the same Smad proteins. In turn, TGF-β1 can induce the expression of activin-A, creating a positive feedback loop mechanism.
Discussion
In this study we demonstrate a marked role for activin-A in cigarette smoke-induced inflammation and COPD using a combination of ex vivo studies on human subjects, in vitro studies on cigarette smoke-exposed primary HBEC cultures and in vivo studies using cigarette smoke-exposed mice. We show for the first time increased levels of activin-A in the lungs of cigarette smoke-exposed mice and patients with COPD. Moreover, we can mimic this in vitro by exposing HBEC cultures to cigarette smoke. Neutralising activin-A with follistatin, its natural inhibitor, in a mouse model of subacute cigarette smoke exposure attenuates inflammation in the BAL fluid.
We demonstrated a higher activin-A mRNA expression in the lungs of current smokers irrespective of airflow limitation, suggesting a cigarette smoke-driven effect rather than an effect of the disease itself. In addition, at the protein level, epithelial activin-A was solely augmented in patients with COPD and not in smokers without airflow limitation, implicating a disease effect. The discrepancy between mRNA and protein levels may be explained by epigenetic mechanisms and/or differences in translational control that contribute to the translation of mRNA into protein. Also in the cigarette smoke-driven murine model of COPD, we revealed increased levels of activin-A in BAL fluid and lung tissue. Importantly, activin-A levels, together with neutrophil and CD8+ T-lymphocyte counts, were still elevated 14 days after final cigarette smoke exposure suggesting that activin-A release is not merely due to an acute cigarette smoke effect. After cigarette smoke cessation, neutrophil counts and activin-A levels dropped more rapidly than CD8+ T-lymphocytes. The persistence of CD8+ T-lymphocytes in the airways can be explained by the recruitment of effector T-cells, which are generated in the draining lymph nodes after stimulation with airway-derived dendritic cells.
Since the increase in activin-A in patients with COPD and cigarette smoke-exposed mice is not accompanied by an upregulation of follistatin there is a relative excess of activin-A, unopposed by follistatin. Similarly, there is evidence from several studies that activin-A is increased in pulmonary diseases such as pulmonary fibrosis and asthma, but also in other non-pulmonary inflammatory disorders such as inflammatory bowel disease and rheumatoid arthritis [16, 20, 27, 28]. Accordingly, in airway epithelium of patients with COPD phosphorylation of Smad2, a downstream mediator of activin-A, was elevated. However, we should keep in mind that Smad2 can also be activated by other ligands of the TGF-β superfamily and that other non-canonical pathways (e.g. nuclear factor-κB) can propagate the activin-A signal [29].
To support the finding that epithelial cells produce activin-A upon cigarette smoke exposure, we exposed primary HBECs in vitro to cigarette smoke. In vitro, it has been shown that cigarette smoke can downregulate the TGF-β pathway in bronchial epithelial cells [30]. However, we demonstrated that mRNA levels of activin-A increase upon cigarette smoke exposure while follistatin expression is hardly influenced. Furthermore, cigarette smoke exposure stimulates the secretion of activin-A and inhibits the secretion of follistatin, resulting in a relative excess of activin-A. Interestingly, activin-A is also increased in cultured airway epithelial cells challenged with human rhinovirus, which is frequently associated with COPD exacerbations [31].
In accordance with the data in patients with COPD, activin-A was increased in the lungs and BAL fluid of mice exposed to cigarette smoke. To elucidate whether activin-A is an innocent bystander or plays an active role in pulmonary inflammation upon cigarette smoke, we blocked activin-A by administering follistatin in cigarette smoke-exposed mice. We demonstrated a significant attenuation of the cigarette smoke-induced increase of CD8+ T-lymphocytes in the BAL fluid, which is the predominant T-cell population in patients with COPD [32]. Strikingly, the secretion of several chemokines (MCP-1 and KC) and cytokines (IL-6 and TNF-α) was reduced in follistatin-treated cigarette smoke-exposed mice compared to the PBS-treated group. Interestingly, the secretion of IL-10, an anti-inflammatory cytokine, is increased upon follistatin treatment in cigarette smoke-exposed mice. Cigarette smoke exposure induced increased regulatory T-cell numbers in the lung and in the draining lymph nodes; however, this was not affected by follistatin treatment (data not shown) [33].
This is the first study exploring the potential of blocking activin signalling to inhibit inflammation in a cigarette smoke-driven murine model. Our in vivo data are in line with experimental data in animal models of other respiratory diseases, showing that the pro-inflammatory effects of activin-A can be blocked in an in vivo setting. Exogenous follistatin reduced the number of macrophages and neutrophils in BAL fluid of a bleomycin-induced rat model of pulmonary fibrosis [34]. Furthermore, neutralisation of endogenous activin-A with ActRIIB-Fc protein reduced the acute lung injury-like pathology in lipopolysaccharide-instilled mice, diminishing neutrophilic inflammation and IL-6 protein levels in BAL [35]. Several reports studied blockade of the activin-A pathway as a treatment for allergic asthma, which resulted in conflicting outcomes. In one study, intranasal administration of follistatin reduced ovalbumin-induced T-helper type-2 immune responses in mediastinal lymph nodes of mice, while another study showed that systemic depletion of activin-A by a neutralising antibody exacerbated ovalbumin-induced asthmatic disease [15, 36]. These latter studies suggest that the route of administration of follistatin may influence the outcome. In our study set-up, systemic administration of follistatin by intraperitoneal injection, it is not possible to differentiate between a local or extrapulmonary effect.
An intriguing aspect of activin-A is that, like TGF-β, it has both pro- and anti-inflammatory activities depending on the type of tissue and injury [37]. A pro-inflammatory activity is suggested by the capability of activin-A to skew the macrophage polarisation towards an M1 phenotype [38]. In contrast, activin-A is known to stimulate the development of FoxP3+ regulatory T-cells [39]. The current hypothesis is that activin-A exerts a pro-inflammatory effect early on in the course of inflammation, but once the inflammatory response has been established, activin-A may stimulate anti-inflammatory effects [40]. Overall, our data suggest that activin-A promotes the pulmonary inflammation after cigarette smoke. Accordingly, when we stimulated HBECs with increasing doses of activin-A for 24 or 48 h, a dose-dependent, but not significant, increase of IL-1β, TGF-β1 and IL-8 mRNA expression was observed, together with an enhanced secretion of IL-8 (fig. E5).
One of the strengths of our study is its translational character. Observations made ex vivo in patients with COPD were confirmed in vitro in HBEC cultures and in vivo in cigarette smoke-exposed mice. A limitation is that only pulmonary inflammation was investigated, which is only one aspect of the complex pathogenesis of COPD. Accumulating data suggest that activin-A is also involved in airway remodelling. In vitro results indicate that activin-A can stimulate proliferation of human lung fibroblasts and smooth muscle cells [41, 42]. Moreover, blocking activin-A inhibited collagen deposition and thickening of the subepithelial smooth muscle layer in two different murine models of allergic asthma [43, 44]. Similarly, it would be helpful to study the relationship between activin-A and peribronchial fibrosis in patients with COPD. In the murine model of COPD it is practically not feasible to study the effect of activin-A silencing on airway remodelling using a pharmacological approach (follistatin or a neutralising antibody), since it is only after 24 weeks of cigarette smoke exposure that airway wall remodelling appears [25]. Interestingly, we demonstrated that follistatin treatment suppresses protein levels of the pro-fibrotic cytokine TGF-β1 in BAL supernatant of cigarette smoke-exposed mice. Since follistatin does not antagonise TGF-β1, the attenuated TGF-β1 levels are probably the result of reduced activin-A based stimulation of TGF-β1 secretion [16]. Importantly, TGF-β1 can also stimulate activin-A expression, inducing a positive feedback loop [16]. In figure 8e we schematically show the effect of cigarette smoke exposure on activin-A signalling in the lungs.
In conclusion, these data demonstrate that activin-A is increased in the airway epithelium of patients with COPD and in the lungs of cigarette smoke-exposed mice. Also, in vitro we confirmed that exposing HBEC cultures to cigarette smoke modulates the delicate balance between activin-A and follistatin. Moreover, our in vivo studies in a cigarette smoke-driven murine model indicate that activin-A contributes to the pathogenesis of cigarette smoke-induced pulmonary inflammation.
Acknowledgments
We would like to thank Greet Barbier, Eliane Castrique, Indra De Borle, Philippe De Gryze, Katleen De Saedeleer, Anouck Goethals, Marie-Rose Mouton, Ann Neesen, Christelle Snauwaert and Evelyn Spruyt (Laboratory for Translational Research in Obstructive Pulmonary Diseases, Dept of Respiratory Medicine, Ghent University Hospital, Ghent, Belgium) for their excellent technical assistance.
Footnotes
For editorial comments see page 954.
This article has supplementary material available from www.erj.ersjournals.com
Support statement: This study was supported by the Concerted Research Action of the Ghent University, Ghent, Belgium (BOF/GOA, 01601009), the Fund for Scientific Research in Flanders (FWO Vlaanderen; G.0195.09 and G.0194.10) and by the Interuniversity Attraction Poles programme (IUAP, P7/30). Part of this work was supported by COST Action BM1201, Developmental Origins of Chronic Lung Disease. F.M. Verhamme is the recipient of a European Respiratory Society Fellowship (STRTF 389-2011). K.R. Bracke is a postdoctoral researcher of the Fund for Scientific Research in Flanders. G.D. Amatngalim is supported by a research grant from Galapagos NV (Mechelen, Belgium).
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
- Received May 13, 2013.
- Accepted October 14, 2013.
- ©ERS 2014
References










