





Abstract
Circulating cardiac troponins are markers of myocardial injury. We sought to determine whether cardiac troponin I (cTnI), measured by a sensitive assay, is associated with disease severity and prognosis in pulmonary arterial hypertension (PAH).
cTnI was measured in 68 patients with PAH diagnostic category 1 in a research-based sensitive immunoanalyser with a lower limit of detection of 0.008 ng·mL−1. The associations between cTnI and PAH severity and clinical outcomes were assessed using Chi-squared and Wilcoxon rank sum tests, Kaplan–Meier analysis and Cox regression models.
cTnI was detected in 25% of patients. Patients with detectable cTnI had more advanced functional class symptoms, a shorter 6-min walk distance, more pericardial effusions, larger right atrial area, and higher B-type natriuretic peptide and C-reactive protein levels. 36-month transplant-free survival was 44% in patients with detectable cTnI versus 85% in those with undetectable cTnI. cTnI was associated with a 4.7-fold increased risk of death related to right ventricular failure or transplant (hazard ratio 4.74, 95% CI 1.89–11.89; p<0.001), even when adjusted individually for known parameters of PAH severity.
Elevated plasma cTnI, even at subclinically detectable levels, is associated with more severe disease and worse outcomes in patients with PAH.
Pulmonary arterial hypertension (PAH) is characterised by luminal obliteration and remodelling of the pulmonary vasculature leading to progressive increments in pulmonary vascular resistance. This leads to overload of the right ventricle, right ventricular failure (RVF) and premature death [1]. Despite impressive advancements in the understanding of the pathobiology of PAH and the introduction of targeted medical therapies, the mortality associated with this condition remains substantial [2]. While guidelines suggest how to tailor different therapeutic approaches according to the severity of the disease [3], risk stratification is difficult and often relies on suboptimal tools such as the assessment of symptoms via the New York Heart Association (NYHA) functional classification. Noninvasive biomarkers of the disease offer tremendous potential to aid difficult therapeutic decisions [4].
The detection of circulating cardiac troponins reflects myocardial necrosis and is diagnostic of acute myocardial infarction in the presence of ischaemic signs and symptoms [5]. In recent years, it has been recognised that elevated cardiac troponins may be detected in other conditions, including stable coronary artery disease [6], left ventricular failure [7], chronic kidney disease [8], sepsis [9] and mixed critical care populations [10]. Elevated cardiac troponins have also been noted in situations of right ventricular overload, both acute, as in acute pulmonary embolism [11], and chronic, as in pulmonary hypertension [12,13]. Mechanisms other that myocardial necrosis may be responsible for this “troponin leak” in these clinical scenarios, even though detectable cardiac troponin is specific for ongoing myocardial damage and generally associated with worse prognosis. In this study, we sought to determine if cardiac troponin I (cTnI), measured with a sensitive assay, is associated with severity and prognosis in diagnostic category 1 PAH patients treated with contemporary PAH-targeted therapy.
MATERIALS AND METHODS
Study population and clinical data collection
Patients were recruited from the Cleveland Clinic Pulmonary Vascular Program (Cleveland, OH, USA). Eligible subjects included patients with PAH diagnostic category 1 according to the updated Dana Point clinical classification [14]. Acute coronary syndromes and advanced renal disease, defined as a serum creatinine >2 mg·dL−1, were further exclusion criteria. Nonconsecutive patients were enrolled after their peripheral blood was collected for study purposes. Venous blood was obtained at the time of right heart catheterisation in 17 incident (newly diagnosed) patients, or during a routine clinical visit where 6-min walk distance (6MWD), NYHA functional class, Doppler echocardiography and brain natriuretic peptide (BNP) measurements were performed in the remaining 51 patients with prevalent disease. Blood was processed and samples were stored in a biobank until biomarker measurements were performed. Time to clinical outcomes was determined from the time of blood sampling. The clinical outcome of interest was transplant-free survival. Events for this outcome were either lung transplantation or death by RVF. Patients who were alive at the end of follow-up and those who died of causes other than RVF were censored. The cause of death was determined by G.A. Heresi by careful review of the medical records and, when needed, discussions with the treating clinician. Clinical events and data were recorded in a prospective fashion in 48 patients and retrospectively in the remaining 20. 6MWD, NYHA functional class, echocardiographic data, and serum creatinine and BNP levels were measured on the same day that study blood sampling took place. Right heart catheterisation data were obtained as close to study blood sampling as possible. The median time between diagnostic right heart catheterisation and blood sampling for study purposes was 240 days. Other data collected included demographic information, traditional cardiovascular risk factors (diabetes mellitus, systemic arterial hypertension and smoking status), coronary artery disease, serum creatinine levels and the use of PAH-targeted therapies. Healthy volunteers were used as controls. This study was approved by the Cleveland Clinic Institutional Review Board (Cleveland Clinic, Cleveland, OH, USA). All study subjects gave written informed consent.
Laboratory measurements
Plasma levels of cTnI were measured using the STAT Troponin I assay (Abbott Laboratories, Inc., Abbott Park, IL, USA) in a research-based immunoanalyser that provides a three-decimal point readout from venous blood samples collected in EDTA tubes. Levels ≥0.03 ng·mL−1 are considered to be diagnostic of myocardial infarction in the right clinical context. We have previously shown that this assay provides highly sensitive analytical measurement of cTnI with a limit of detection of 0.008 ng·mL−1 [15]. However, the assay detected cTnI levels between 0.001 and 0.008 ng·mL−1 in 1,291 subjects out of a total of 3,828 stable patients undergoing an elective cardiac evaluation. Importantly, cTnI levels between 0.001 and 0.008 ng·mL−1 were associated with an increased risk of death, myocardial infarction or stroke compared with levels <0.001 ng·mL−1 [15]. This assay has also been shown to be comparable with other high-sensitivity assays [16]. Thus, for the purpose of this study, we defined detectable cTnI as any level >0.001 ng·mL−1. In the PAH cohort, we also measured high-sensitivity C-reactive protein (CRP) and high-density lipoprotein cholesterol (HDL-C) using the Abbott ARCHITECT platform. We also measured cardiac troponin T (cTnT) with the Elecsys Troponin T assay (Roche Diagnostics, Indianapolis, IN, USA) in 62 out of the 68 patients. This assay provides a two-decimal readout with a limit of detection of 0.01 ng·mL−1
Statistical analysis
Demographic, clinical and laboratory data were summarised as frequency (%) and mean±sd. cTnI levels were compared between PAH patients and controls by means of Wilcoxon rank sum and Chi-squared tests. Patient groups defined by cTnI status were compared with respect to categorical study variables using Chi-squared and Fisher's exact test, and with respect to quantitative and ordinal variables using Wilcoxon rank sum tests. Correlations between cTnI and variables of interest were estimated by the Spearman correlation coefficient.
The association between transplant-free survival and cTnI was assessed using log-rank tests, with Kaplan–Meier estimation used to describe the likelihood of the outcomes during the follow-up period. Patients who died of causes other than RVF were censored for this analysis. Cox regression models were used to provide estimates of hazard ratios and 95% confidence intervals for transplant-free survival with respect to their associations with cTnI. Cox regression was also used in a multivariable fashion to explore the covariate-adjusted associations between transplant-free survival and cTnI. We adjusted for selected covariates individually, as the sample size and number of events did not allow for building models with multiple covariates. Results for covariate adjusted associations were summarised using Wald p-values and covariate-adjusted hazard ratio estimates. Analyses were performed using R version 2.8.1 (R Development Core Team 2011; R Foundation for Statistical Computing, Vienna, Austria) [17].
RESULTS
Study population and cTnI levels
We studied 68 PAH patients (aged mean±sd 47±13 yrs, 91% female) and 29 controls (aged 35±6 yrs, 75% female). PAH categories were as follows: idiopathic PAH (n=41); heritable PAH (hereditary haemorrhagic telangiectasia; n=1); and associated PAH (n=26; 20 connective tissue disease, three congenital heart disease, two chronic haemolytic anaemia and one portopulmonary). At the time of study enrolment, 51 (75%) patients were receiving PAH-targeted therapies: intravenous prostacyclin (n=15); phosphodiesterase inhibitor (PDE5i) (n=7); endothelin receptor antagonist (ERA) (n=4); i.v. prostacyclin plus ERA (n=13); i.v. prostacyclin plus PDE5i (n=3); ERA plus PDE5i (n=5); and i.v. prostacyclin plus ERA plus PDE5i (n=4). The 17 patients naive to therapy at enrolment (25% of the cohort) were eventually treated as follows: i.v. prostacyclin (n=2); PDE5i (n=4); i.v. prostacyclin plus PDE5i (n=4); ERA plus PDE5i (n=3); i.v. prostacyclin plus PDE5i (n=2); and i.v. prostacyclin plus ERA plus PDE5i (n=2). Table 1 details functional, echocardiographic and haemodynamic characteristics of the PAH cohort. No PAH patient had diabetes, 15 (22%) had systemic hypertension and 27 (40%) were either former or active smokers. Three (4%) patients had coronary artery disease, none of whom had detectable cTnI.
17 (25%) PAH patients had detectable cTnI levels (>0.001 ng·mL−1), compared with one (3.4%) control (p=0.011). Two (2.9%) patients had cTnI levels between 0.001 and 0.008 ng·mL−1. Nine (13.2%) patients had cTnI levels between 0.009 and 0.029 ng·mL−1, as did the one control with detectable cTnI. Six (8.8%) patients had levels >0.030 ng·mL−1, none of whom had coronary artery disease. In patients with detectable cTnI, mean±sd levels were 0.037±0.009 ng·mL−1, with a range of 0.003–0.140 ng·mL−1. The detection rate for cTnT was only four (6.5%) out of 62 (p=0.006 for the comparison with the 25% detection rate of cTnI). All patients with detectable cTnT also had a detectable cTnI. Conversely, 12 patients with detectable cTnI had undetectable cTnT.
cTnI and PAH severity
cTnI concentrations had significant positive correlations with NYHA functional class, right atrial (RA) area measured with echocardiography, and BNP and CRP levels (table 2). There was a borderline positive correlation with right atrial pressure (Pra) (r=0.24, p=0.06). Negative correlations with 6MWD, mixed venous oxygen saturation and HDL-C levels (r= -0.42) were observed (table 2).
Patients with detectable cTnI were more likely to have associated PAH, more advanced functional class symptoms and more pericardial effusions; they also had shorter 6MWD, higher Pra, larger RA area, higher BNP and CRP levels, and lower HDL-C concentrations (table 1). Both subgroups were similar in age, sex, cardiac frequency and systemic blood pressure, as well as in the proportion of subjects with systemic hypertension and tobacco use.
Clinical outcomes
22 (32.4%) patients died and two (2.9%) underwent lung transplantation. Nine (53%) out of 17 patients with detectable cTnI died, compared with 13 (25%) out of 51 with undetectable cTnI (p=0.041). All of the nine deaths in patients with detectable cTnI were due to RVF. Among the 13 deaths with undetectable cTnI, three were secondary to severe infections (two pneumonias and one catheter-related bloodstream infection), one was related to pancreatic cancer and one could not be ascertained; the remaining eight deaths were secondary to RVF. There were no sudden deaths in either group. The proportion of transplant or death related to RVF in patients with undetectable cTnI was 17.6% (nine out of 51) compared with 58.8% (10 out of 17) in patients with detectable cTnI (p=0.003). 15 (26%) out of 58 patients with undetectable cTnT died of RVF at the end of follow-up, compared with three out of four patients (75%) with detectable cTnT (p=0.07).
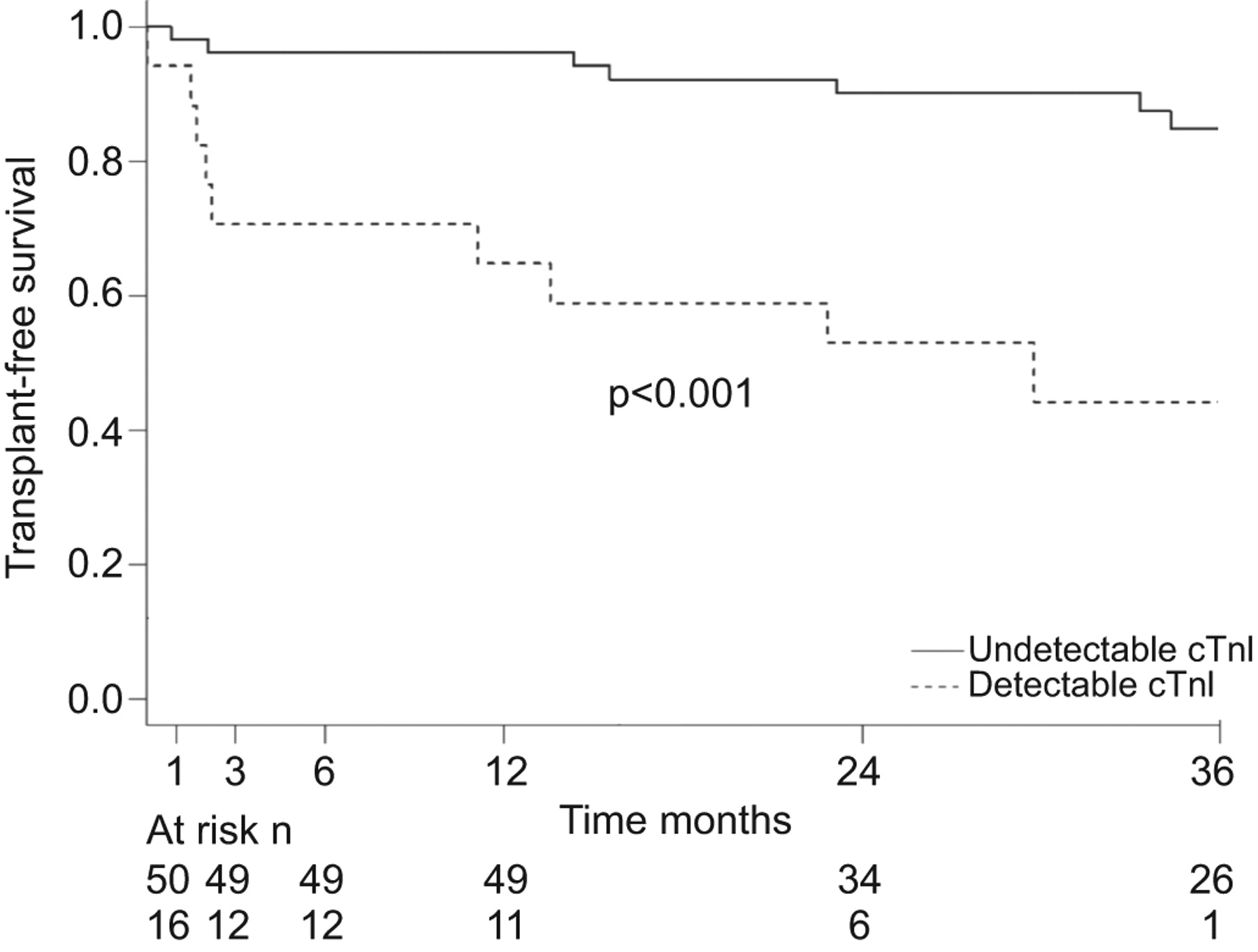
Detectable cTnI was associated with a 4.7-fold increased risk of death related to RVF or transplant: hazard ratio (HR) 4.74, 95% CI 1.89–11.89 (p<0.001) (fig. 1 and table 3). In patients with detectable cTnI, 12-, 24- and 36-month transplant-free survival was 64.7, 52.9 and 44.1% respectively, compared with 96.1, 90 and 84.7% in patients with undetectable cTnI. Table 3 shows that age, type of PAH, NYHA functional class, 6MWD, pericardial effusion, Pra and BNP were associated with the risk of death by RVF or transplant. We also found prognostic values for HDL-C and creatinine, but not for CRP. In this cohort, there was no association between prevalent versus incident cases and different PAH-targeted therapies with clinical outcomes (table 3). The association between cTnI and transplant-free survival remained unchanged when adjusted individually for age, PAH subcategory (idiopathic versus associated), NYHA functional class, 6MWD, the presence of pericardial effusion, Pra, cardiac index, the use prostacyclin therapy, and HDL-C, CRP and creatinine concentrations (all p<0.05) (table 4). Adjustment for the presence of systemic hypertension, coronary artery disease, statin therapy and body mass index also did not affect this association. There was a significant interaction between cTnI and BNP (p=0.006). Patients with both low BNP levels and undetectable cTnI had longer transplant-free survival, while those with both detectable cTnI and supra-median BNP had the worst outcomes, but small numbers of patients in each group do not allow for firm conclusions. When analysing all-cause mortality and transplantation, detectable cTnI was still highly predictive of worse outcomes (HR 3.21, 95% CI 1.40–7.37; p=0.004). When excluding the two patients with cTnI levels between 0.001 and 0.008 ng·mL−1, detectable cTnI was still highly associated death by RVF (HR 4.07, 95% CI 1.54–10.78; p<0.001) (see online supplementary figure 1 and table 1).
{kind=link}
Transplant-free survival according to cardiac troponin I (cTnI) status. Patients with detectable cTnI had a 4.7-fold higher risk of right ventricular-related death or lung transplantation compared to patients with undetectable cTnI.
DISCUSSION
The main finding of our study is that cTnI detected by a sensitive assay even at subclinical levels is associated with more severe disease and worse transplant-free survival in patients with PAH. The current study uses a sensitive troponin assay and provides the largest cohort with a homogenous category 1 PAH population treated with contemporary PAH-targeted therapies for the longest follow-up time.
The elevated pulmonary vascular resistance that characterises PAH leads to increase intramural right ventricular tension and eventually decreased cardiac output, which in turn can lead to lower systemic pressures. This situation is conducive to right ventricular myocardial ischaemia and necrosis, which may account for the troponin leak observed in a quarter of our patients. Our data support this notion, as we observed more right heart chamber dilation and dysfunction in patients with detectable cTnI. Mechanisms other than myocardial ischaemia are also possible. We found a positive correlation between cTnI and CRP levels, suggesting that inflammatory mechanisms may play a role. In the present study, a 1 sd increase in CRP levels was not associated with death by RVF (table 3). This contrasts with the report by Quarck et al. [18] showing decreased survival with CRP levels above the upper limit of normal of 5 mg·L−1. We have previously reported a similar association with regards to clinical worsening, but not survival [19]. While differences in the statistical methods used may account for these discrepancies, the relationship between CRP and clinical outcomes deserves further study.
The previous two studies of cardiac troponin in pulmonary hypertension are limited by a number of issues: the use of a lower-sensitivity assay [12]; the inclusion of heterogeneous populations with pulmonary hypertension secondary to chronic thromboembolic disease and interstitial lung disease [12,13]; low numbers of category 1 PAH subjects [13]; short-term follow-up with small numbers of clinical events [13]; and noncontemporary management of PAH [20]. In the present study, we used an assay that provides highly sensitive analytical measurement of cTnI with a three-decimal point readout, which has been found to be comparable with other high-sensitivity assays [16]. We detected cTnI in 25% of the patients with the sensitive assay, and cTnT in 6.5% of patients, compared with a detection rate of 13–14% for cTnT in previous studies in Poland [12,21], and 4.3% in a study of chronic thromboembolic pulmonary hypertension by Lankeit et al. [22]. Our lower detection rates for cTnT may be explained by the inclusion of a majority of patients receiving PAH-targeted therapies. Our findings for cTnT mirror the results of Lankeit et al. [22] (very low detection rate and poor outcomes in all patients with detectable cTnT) and suggest that using an assay with a two-decimal point readout for cTnT does not provide enough prognostic sensitivity. We have shown that the association between cardiac troponins and poor outcomes extends to lower troponin levels not detected with assays in current clinical use.
We report on a cohort that includes only diagnostic category 1 PAH, according to the recent classification [14], who were treated with contemporary PAH management [20]. The latter may explain the 52.9% 24-month survival rate in our cohort with detectable cTnI, compared with 29% in the report by Torbicki et al. [12] published in 2003. Remarkably, even with improved outcomes, cTnI was predictive of transplant-free survival in our cohort. The sensitive assay used for this study, which allows for the detection of subclinical levels of cTnI, may have contributed to our ability to show this predictive ability. A recent French study failed to show any association between cTnI and survival in patients with acutely worsened PAH, with the authors noting that more sensitive assays for troponin measurements should be used [23].
Detectable cTnI was associated with more severe PAH, as suggested by more advanced functional class symptoms, more pericardial effusions, shorter 6MWDs, higher Pra, larger RA area, higher BNP and CRP levels, and lower HDL-C concentrations. The detection of cTnI by our sensitive assay carried a 4.7-fold increase risk of death related to RVF or transplant, with 36-month transplant-free survival of 44.1%, compared with 84.7% in patients with undetectable cTnI. cTnI was predictive of worse outcomes when adjusted individually for several covariates known to influence survival in PAH, including age, idiopathic versus associated PAH, functional class, 6MWD, pericardial effusion, Pra, cardiac index, creatinine, CRP and HDL-C. As they both reflect right ventricular strain, it is not surprising that we found a strong correlation between cTnI and BNP. Furthermore, we found a significant interaction between these markers (data not shown). The addition of cTnI to BNP may allow for better risk stratification.
The study of a relatively small and heterogeneous PAH population, including idiopathic disease and cases associated with connective tissue disease, constitutes a limitation of our study, and one that is common to single-centre biomarker investigations. Nevertheless, when adjusted for PAH category (idiopathic versus associated disease), cTnI remained associated with worse outcomes. Another limitation of this study is the inclusion of incident and prevalent cases, where 75% of the patients were receiving PAH-targeted therapies when cardiac troponins were measured. While this may have biased the results and also explains the very low detection rate for cTnT, it is noteworthy that a more sensitive troponin assay still provided prognostic information in patients receiving contemporary PAH therapies. Before a novel biomarker can be recommended for widespread clinical use, it needs to be evaluated in a rigorous fashion [24]. While biological plausibility certainly exists for the use of circulating troponins in PAH, and the available evidence suggests a potential role, further studies with larger numbers of patients are needed to confirm their utility. For example, a recent systematic review of the literature suggested that troponins may not be discriminative enough to be used in the risk stratification of patients with normotensive acute pulmonary embolism [25]. As cardiac troponin assays become more sensitive, cut-offs associated with meaningful clinical outcomes will have to be defined to avoid diagnostic confusion. For instance, in patients suspected of having acute myocardial infarction, the higher-sensitivity assays improve diagnostic sensitivity, but at the expense of lower specificity [16,26]. Recently, cTnT has been detected in 25% of the general population [27] and 66% in people aged >65 yrs [28] using highly sensitive assays with a limit of detection of 0.003 ng·mL−1. Rates of detection are much lower in females and young people [27], which is relevant to patients afflicted with PAH.
In summary, using a sensitive assay, we found a 25% detection rate for cTnI in a cohort of category 1 PAH patients. Detectable cTnI was associated with more severe PAH and worse clinical outcomes. The measurement of circulating cTnI with a sensitive assay may help clinicians to identify high-risk patients who could benefit from more aggressive therapies.
Footnotes
For editorial comments see page 799.
This article has supplementary material available from www.erj.ersjournals.com
Statement of Interest
Statements of interest for G.A. Heresi, W.H.W. Tang, S.L. Hazen and R.A. Dweik can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received April 19, 2011.
- Accepted August 10, 2011.
- ©ERS 2012
REFERENCES










