





Abstract
The expression of nuclear factor (NF)-κB is an indicator of cellular activation and of inflammatory mediator production. The aim of the present study was to characterise the expression and localisation of p65, the major subunit of NF-κB, in the bronchial mucosa of patients with chronic obstructive pulmonary disease (COPD), and to examine the relationship between p65 expression and disease status.
Bronchial biopsies were obtained from 14 smokers with COPD, 17 smokers with normal lung function and 12 nonsmokers with normal lung function. The number of p65 positive (+) cells was quantified by immunohistochemistry and the expression of p65 in bronchial biopsies from the three groups was examined by Western blotting (WB).
Smokers with normal lung function and patients with COPD had increased numbers of p65+ cells in the epithelium and increased p65 nuclear expression. In COPD patients the number of epithelial p65+ cells correlated with the degree of airflow limitation. WB analysis showed an increase in p65 in smokers with normal lung function and COPD patients (p<0.05).
Bronchial biopsies in smokers with normal lung function and chronic obstructive pulmonary disease patients show increased expression of p65 protein, predominantly in the bronchial epithelium. Disease severity is associated with an increased epithelial expression of nuclear factor-κB.
This study was supported by the Associazione per la Ricerca e la Cura dell'Asma (ARCA, Padua, Italy), Salvatore Maugeri Foundation, IRCCS, “Ricerca Corrente” and Glaxo-Wellcome (UK).
Chronic obstructive pulmonary disease (COPD) can be defined as chronic irreversible airflow limitation confirmed by spirometry 1–3 and it is characterised by a slowly progressive and irreversible deterioration in lung function 1–3. Cigarette smoking is the major risk factor for the development of COPD and cigarette smokers constitute over 90% of COPD patients in developed countries 1–3. Recent studies performed on bronchial biopsies from patients with COPD reported an increased infiltration of T-lymphocytes (CD3+ and CD8+ cells), macrophages 4–6 and increased levels of the C-C chemokine receptor 5 in mild-to-moderate COPD 7. Furthermore, the inflammatory response in COPD is associated with increased expression of inflammatory proteins, such as cytokines, chemokines 8 and adhesion molecules 5, 9, 10. The inflammatory changes that occur in COPD are also seen in cigarette smokers without COPD, but to a lesser extent 9.
The inducible genes that encode these inflammatory proteins are regulated by transcription factors, which bind to the regulatory (promoter) regions of these genes to modulate their rate of transcription 10. Although many transcription factors are probably involved in the regulation of these inflammatory genes, one, nuclear factor-κB (NF-κB) appears to be of particular importance 11. The genes for a number of cytokines and other mediators which have been reported to be involved in the inflammatory process of the airways in COPD, including interleukin (IL)-1 12, IL-6 13, IL-8 14–16, monocyte chemoattractant protein (MCP)-1 17, tumour necrosis factor (TNF)-α 18, endothelin-1 19, secretory leucocyte proteinase inhibitor 20 and intercellular adhesion molecule (ICAM)-1 21, 22, are all regulated by NF-κB 23–25.
The activated form of NF-κB is a heterodimer, which usually consists of two subunits, p65 (RelA) and p50. In resting cells NF-κB is retained in the cytoplasm as a result of binding to inhibitory proteins (IκB). When the cell is appropriately stimulated, degradation of IκB uncovers nuclear localisation signals on p65, so it is rapidly transported into the nucleus where it binds to specific κB recognition elements in the promoter regions of target genes 23. Many extracellular stimuli, including IL-1β, TNF-α, oxidants and lipopolysaccharide activate NF-κB in many cells including epithelial cells and T-lymphocytes 10.
A previous study has suggested that cellular expression of p65 protein may be related to disease status and T-lymphocyte infiltration of the bronchial mucosa in patients with asthma 26. In the present study the authors identified and quantified the number of cells expressing the p65 protein in the epithelium and submucosa of bronchial biopsies from COPD patients, and for comparison in a control group of smokers and nonsmokers with normal lung function. The percentage of T-cell subsets (CD4+ and CD8+) co-expressing p65 protein in the submucosa of bronchial biopsies from COPD patients was also quantified.
Methods
Subjects
Biopsies from 43 subjects were examined by immunohistochemistry, 31 were smokers and 12 were nonsmokers with normal lung function. The severity of airflow limitation was staged using the criteria of the Global Initiative for Chronic Obstructive Lung Disease 1: 14 smokers had mild-to-moderate COPD (forced expiratory volume in one second/forced vital capacity (FEV1/FVC) ratio <70% predicted and FEV1 >30% pred) and 17 had normal lung function (healthy smokers, FEV1/FVC ratio >70% pred) (table 1⇓).
Subject characteristics
Six control nonsmokers, eight healthy smokers and five COPD patients (mean±sd: FEV1 % pred: 107±8 versus 94±9 versus 72±15 (p<0.05 compared with control smokers and control nonsmokers, respectively) were used for Western-blot analysis of p65 expression in the bronchial biopsies.
None of the study subjects had suffered a recent exacerbation, defined as increased dyspnoea associated with a change in quality and quantity of sputum that would have led them to seek medical attention during the month preceding the study. All subjects were free of acute upper respiratory-tract infections and none had received glucocorticoids, theophylline or antibiotics within the preceding month. All subjects were nonatopic (i.e. they had negative skin tests for common allergen extracts) and had no past history of asthma or allergic rhinitis.
Each subject underwent an interview, chest radiograph, electrocardiogram, routine blood test, skin tests with common allergen extracts, and pulmonary function tests between 2–5 days before bronchoscopy. The study conformed to the Declaration of Helsinki, and informed consent was obtained from each subject. Bronchial biopsies were performed according to the local ethics committee guidelines.
Lung function tests and volumes
Pulmonary function tests included measurements of FEV1 and FEV1/FVC under baseline conditions in all the subjects examined (6200 Autobox Pulmonary Function Laboratory; Sensormedics Corp., Yorba Linda, CA, USA). The predicted normal values used were those from the Communité Européenne du Carbon et de l′Acier 27. In order to assess the reversibility of airway obstruction, the FEV1 measurement in the group of subjects with FEV1 ≤80% was repeated 20 min after the inhalation of 200 µg of salbutamol.
Fibreoptic bronchoscopy, collection and processing of bronchial biopsies
Nonsmokers and smokers with normal lung function and COPD patients attended the bronchoscopy suite at 08.30 h after having fasted from midnight and were pretreated with atropine (0.6 mg i.v.) and midazolam (5–10 mg i.v.). Oxygen (O2) (3 L·min−1) was administered via nasal prongs throughout the procedure and O2 saturation was monitored with a digital oximeter. Using local anaesthesia with lidocaine (4%) to the upper airways and larynx, a fibreoptic bronchoscope (Olympus BF10 Key-Med, Southend, UK) was passed through the nasal passages into the trachea. Further lidocaine (2%) was sprayed into the lower airways, and four bronchial mucosal biopsy specimens were taken from segmental and subsegmental airways of the right lower and upper lobes using size 19 cupped forceps. Bronchial biopsies for immunohistochemistry were processed as described later. Bronchial biopsies for Western-blot analysis were immediately placed on ice and processed as previously described 28.
Immunohistochemistry
Biopsy specimens were gently extracted from the forceps and processed for light microscopy as previously described 6, 7. Two samples were embedded in Tissue Tek II optimal cutting temperature (OCT) (Miles Scientific, Naperville, IL, USA), frozen within 15 min in isopentane precooled in liquid nitrogen, and stored at −80°C. The best frozen sample was then oriented and 6-µm thick cryostat sections were cut for immunohistochemical light-microscopy analysis. Two sections at an interval of 100 µm were then appropriately stained with immunohistochemical methods. The following panel of antibodies were used to identify T-lymphocytes, T-lymphocyte subsets (CD4+ and CD8+ cells) and NF-κB p65 protein: mouse anti-CD3 antigen (M756; Dako, Ely, UK) for total T-lymphocytes, mouse anti-CD4 antigen (M716; Dako) for T-helper, mouse anti-CD8 antigen (M707; Dako) for T-suppressor/cytotoxic, and rabbit anti-p65 (sc-372, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for NF-κB p65 protein. Primary antibodies were determined as previously described 6, 7. In brief, after blocking nonspecific binding sites using serum derived from the same animal species as the secondary antibody, mouse primary antibodies were applied at optimal dilutions in tris-(hydroxymethyl)-aminomethane (TRIS)-buffered saline (0.15 M saline containing 0.05 M TRIS-hydrochloric acid at pH 7.6) and incubated (1 h) at room temperature in a humidified chamber. Antibody binding was demonstrated with the use of an alkaline phosphatase anti-alkaline phosphatase system (Dako APAAP Kit System K 670) and fast-red substrate. Application of the rabbit polyclonal antibody (1 h) was preceded by incubation with 3% H2O2 for 20 min followed by incubation in avidin (20 min) and biotin (20 min) blocking solutions (SP-2001, Vectastain Kit) and blocking serum. The rabbit polyclonal antibody was than revealed by incubation in a biotinylated swine anti-rabbit antibody (E431, Dako, 30 min), ABC complex HRP (K377, Dako, 30 min) and 3,3′ diaminobenzidine tetrahydrochloride (S3000, Dako, 15 min). Double-staining was performed with the rabbit anti-p65 protein and mouse anti-CD4 and CD8-antigens as primary antibodies (1 h) followed by incubation with swine anti-rabbit horseradish peroxidase conjugated (P399; Dako) and goat anti-mouse alkaline phosphatase conjugated (A2179; Sigma) secondary antibodies (1 hr) revealed by incubation in 3,3′ diaminobenzidine tetrahydrochloride (S3000, Dako, 15 min) and fast-red substrates (APAAP Kit system K699, Dako, 20 min). Control slides were included in each staining run using human tonsil as a positive control for immunostaining of total T-lymphocytes (CD3 antigen), T-lymphocyte subsets. CD4+, CD8+ cells, p65+ cells, as well as double-stained cells were quantified in the area 100 µm beneath the epithelial basement membrane in several nonoverlapping high power fields until all the available area was covered. The final result, expressed as the number of positive cells per square millimetre, was calculated as the average of all the cellular counts performed in both slides of each biopsy. Double stained p65+/CD4+, p65+/CD8+ were quantified in the same area and the final result was expressed as percentage of CD4+ and CD8+ cells, respectively. The p65+ cells were also quantified in the well-preserved epithelium (constituted by columnar and basal epithelial cells) and expressed as number of p65+ cells per millimetre of epithelial length. A mean±sd of 0.750±0.350 mm of epithelium was analysed in COPD patients and control subjects. Counting was performed by a colleague in a blinded manner.
Light-microscopic analysis was performed at a magnification of x900 for quantification of T-lymphocytes, T-lymphocyte subsets, p65 protein, and double-stained cells in the epithelium and submucosa. Morphometric measurements were performed with a light microscope (Leitz Biomed, Leica Cambridge, UK) connected to a video recorder linked to a computerised image system (Quantimet 500 Image Processing and Analysis System, Software Qwin V0200B, Leica).
For confocal microscopy sections were stained with p65 (1:50 dilution, Santa Cruz Biotechnology) and visualised using a rhodamine-conjugated secondary antibody exactly as previously described 29. Nuclei were visualised using Vectashield-mounting medium with 4,6-diamino-2-phenylinodole (DAPI) (Vector Laboratories) staining and the cover glass sealed with mounting medium (distrene, plasticiser, xylene (DPX)). Slides were viewed using confocal microscopy. Confocal-scanning laser microscopy images were collected with a Lieca confocal microscope, equipped with a 488/514 nm dual-band argon-ion laser. An oil-immersion objective was used. Images were collected using TCSNT software (Leica).
Western-blot analysis for p65
Whole cell proteins were extracted from bronchial biopsies as previously described 28. In brief, bronchial biopsies were resuspended with mechanical disruption in 30–50 µL of 1X reporter-lysis buffer (Promega, Southampton, UK) with a protease inhibitors cocktail (Roche Molecular Biochemicals, Lewes, UK) immediately frozen to −70° C and thawed after at least 60 min. Protein concentration was measured in the supernatant by the Bradford method according to the manufacturer's instructions (Bio-Rad Laboratories, Hemel Hempstead, UK). At least 30 µg·lane−1 of whole-cell proteins were subjected to a 10% sodium dodecylsulphate (SDS)-polyacrylamide gel electrophoresis, and transferred to nitrocellulose filters (Hybond-ECL, Amersham Pharmacia Biotech) by blotting. Filters were blocked using 5% nonfat dry milk before incubation with rabbit anti-p65 antibody (Santa Cruz Biotechnology). p65 was detected using an antirabbit antibody conjugated to horseradish peroxidase (Dako) at dilution of 1:4000 and immunocomplexes visualised using enhanced chemiluminescence (ECL) as recommended by the manufacturer (Amersham Pharmacia Biotech).
As an internal control each filter was reprobed with an antihuman actin antibody (Santa Cruz Biotechnology).
The 43 kDa (actin) or 65 kDa (p65) bands were quantified using densitometry with Grab-It and GelWorks software (UVP, Cambridge, UK) and expressed as the ratio with the corresponding actin optical-density value of the same lane.
Data analysis
Group data were expressed as mean±sd. Differences between groups were analysed using analysis of variance (ANOVA) for functional data, and the Kruskal-Wallis test for morphological data. When the differences were significant, the ANOVA test was followed by an unpaired t-test for comparison between groups and the Kruskal-Wallis test was followed by the Mann-Whitney U-test for comparison between groups. Correlation coefficients were calculated using Spearman's rank method. Probability values of p<0.05 were considered as significant. Data analysis was performed using the Stat View SE Graphics program (Abacus Concepts Inc., Berkeley, CA, USA).
Results
Clinical findings
Subject characteristics are reported in table 1⇑. The three groups of subjects examined were similar with regard to age. Smoking history was similar in COPD patients and healthy smokers. As expected from the selection criteria, the values of FEV1 (% pred) and FEV1/FVC (%) were significantly different in the group with COPD compared to both healthy smokers and control nonsmokers (for overall groups, ANOVA test: p<0.0001 for FEV1% and FEV1/FVC% values).
p65 immunohistochemistry in the bronchial epithelium
The total number of p65 immunoreactive cells in the bronchial epithelium was significantly increased in smokers with normal lung function (215±55 cells·mm−1) and COPD patients (279±95 cells·mm−1) compared with nonsmokers with normal lung function (143±37 cells·mm−1; overall groups: p=0.0007; figs 1–2⇓⇓⇓⇓).

Photomicrographs showing the bronchial epithelium of a a) control nonsmoker, b) healthy smoker and c) patient with chronic obstructive pulmonary disease (COPD) immunostained for identification of p65+ cells. Insets show the bronchial epithelium at a higher magnification. E: epithelium. Arrows indicate representative positively stained cells. Results are representative of those from 12 nonsmokers, 17 healthy smokers and 14 subjects with COPD. Internal scale bars=30 μm and 12 μm for insets.

p65 positive cells in the a) epithelium and b) submucosa of subjects with chronic obstructive pulmonary disease (COPD; └), control smokers ( ) and control nonsmokers (□). The results are expressed as the mean±sd positive cells per millimetre of epithelium. Statistical analysis: Mann-Whitney U-test for comparison between groups. Nonsmokers: n=12; healthy smokers: n=17; COPD: n=14. *: p<0.05.
) and control nonsmokers (□). The results are expressed as the mean±sd positive cells per millimetre of epithelium. Statistical analysis: Mann-Whitney U-test for comparison between groups. Nonsmokers: n=12; healthy smokers: n=17; COPD: n=14. *: p<0.05.

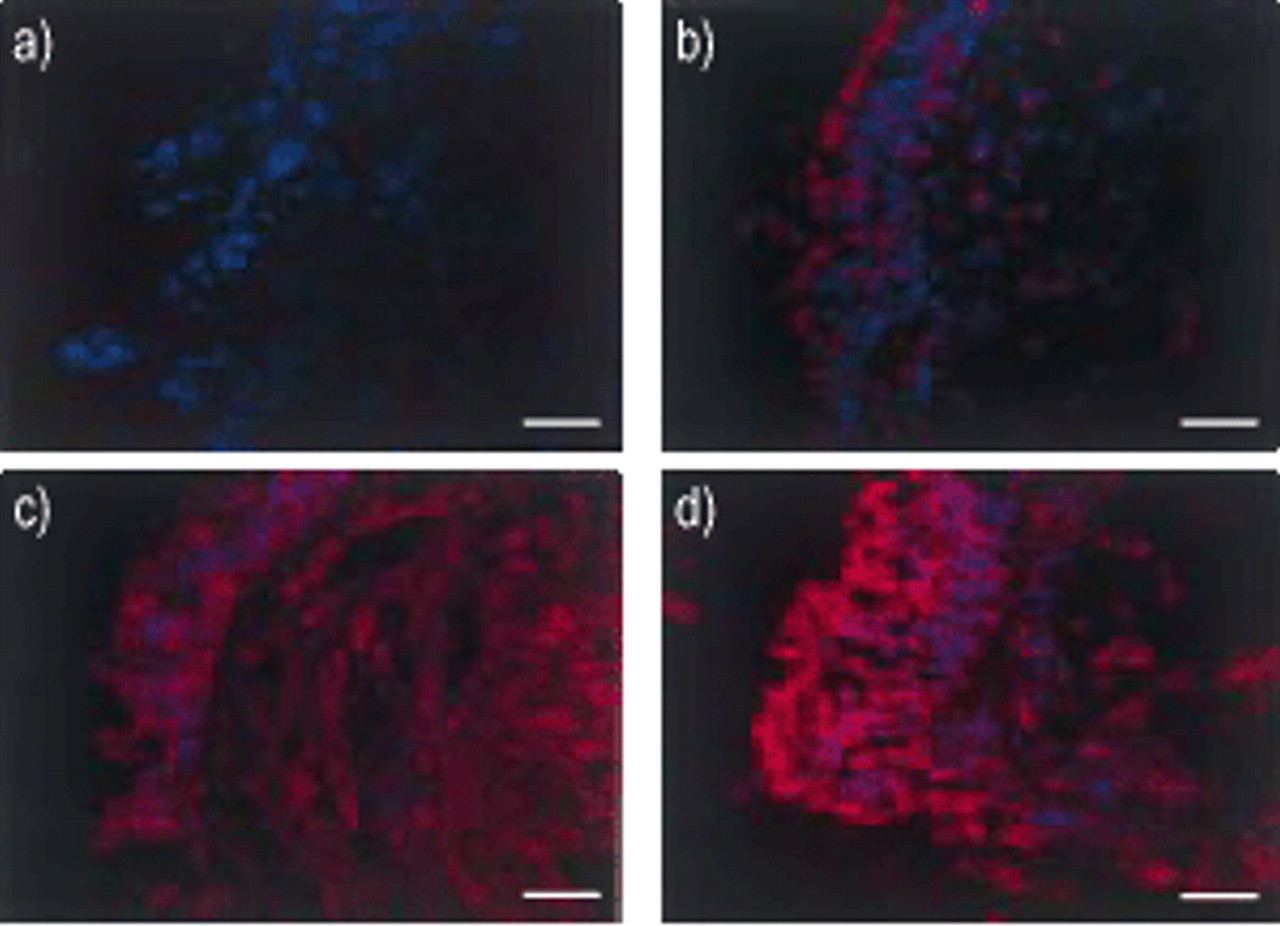
Confocal microscopic analysis of p65 staining (red) in epithelial cells. a) Negative control, b) control nonsmoker, c) smoker and d) chronic obstructive pulmonary disease patients. Nuclei are stained blue by 4,6-diamino-2-phenylindole (DAPI). Internal scale bar=12 μm.

The intensity of p65 cells is reduced with increasing nuclear p65 localisation. Results presented as mean±sem. **: p<0.01, ***: p<0.001.
In the bronchial epithelium there was no significant difference between smokers with normal lung function and COPD patients in the total number of p65 immunoreactive cells (figs 1–4⇑⇑⇑⇑).
p65 immunohistochemistry in the submucosa
The total number of p65-immunoreactive cells in the submucosa was not statistically different between control nonsmokers, smokers with normal lung function and COPD patients (fig. 2⇑⇑⇑⇑).
p65 nuclear staining
Overlaid images of red p65 staining and blue (DAPI) nuclear staining showed less intense blue colouration in the smokers and subjects with COPD indicating colocalisation (fig. 3⇑). p65 nuclear staining was increased in healthy smokers (70±2, p<0.01) and subjects with COPD (76±2, p<0.001) compared to normal control subjects (35±12, figs 3–4⇑). The intensity of staining was also scored on a 1–4 intensity scale and confirmed the light-microscopy data. Normal subjects (1.5±0.5) had less intense p65 staining compared with healthy smokers (2.5±0.3, p<0.05) and subjects with COPD (2.8±0.2, p<0.05).
CD3, CD4 and CD8 immunohistochemistry in the bronchial submucosa
The number of CD3+ and CD8+ cells was significantly increased in the submucosa of smokers with normal lung function and COPD patients compared with nonsmokers with normal lung function (CD3+: 334±117 versus 539±238 versus 556±229, p<0.05 compared with nonsmokers; CD4+: 182±75 versus 214±152 versus 294±165; CD8+: 144±65 versus 334±175 versus 275±98, p<0.05 compared with nonsmokers, respectively for nonsmokers, smokers and COPD). Double staining in the bronchial submucosa of COPD patients for identification of CD4+ and CD8+ cells co-expressing the p65 protein showed that approximately one-third of both T-cell subsets co-expressed p65 (fig. 5⇓).

Photomicrographs showing the bronchial mucosa of patients with a) chronic obstructive pulmonary disease (COPD) immunostained for identification of p65+ (brown stain)/CD4+ (red stain) cells and b) p65+(brown stain)/CD8+ (red stain) cells, in the submucosa (arrows). Results are representative of those from 12 nonsmokers, 17 healthy smokers and 14 subjects with COPD. COPD patients: n=6. Mean±sem: 31±14% for p65+CD4+/CD4+; 33±8 for p65+/CD8+.
Western-blot analysis for p65 in bronchial biopsies
Quantification of total p65 protein in the bronchial biopsies of six nonsmokers, eight smokers with normal lung function and five COPD patients by Western-blot analysis showed a significant increase of the p65/actin ratio in the smokers with normal lung function (1.01±0.65) and COPD patients (0.66±0.34) compared with control nonsmokers with normal lung function (0.25±0.20; p<0.05) (fig. 6⇓). There was no significant difference between smokers with normal lung function and COPD patients in the total amount of p65 in the bronchial biopsies (fig. 6⇓).

a) Western blot of p65 protein expression (with β-actin controls) in bronchial biopsies of control nonsmokers (normals), healthy smokers (smokers) and chronic obstructive pulmonary disease (COPD) patients. b) p65:actin ratio. Results are representative of experiments from six normal subjects, eight smokers and five COPD subjects in each group. *p<0.05.
Correlations between morphological and lung function data
When analysed globally across all smokers (either with normal lung function or COPD patients) the number of p65-immunoreactive cells in the epithelium was inversely and significantly correlated with the FEV1 (% pred) (r=−0.43, p<0.05) and FEV1/FVC ratio (r=−0.50, p<0.05) (fig. 7⇓). When the analysis was restricted to the patients with airflow limitation (COPD patients), these correlations were maintained (r=−0.78, p<0.05 for FEV1%; r=−0.75, p<0.05 for FEV1/FVC%) (fig. 7⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Regression analysis between forced expiratory volume in one second (FEV1) % predicted (a) and b)), FEV1/forced vital capacity (FVC) % (c) and d)) values and numbers of total p65+ cells in the epithelium of all smokers (a) and c)) and smokers with chronic obstructive pulmonary disease alone (b) and d)) (Spearman's rank correlations). a) r=−0.43; p=0.045; b)r=−0.78, p=0.013; c) r=−0.50; p=0.017; d) r=−0.75, p=0.017.
No other significant correlations between the morphological and functional data or between morphological parameters in the whole-subjects group or in subjects with airflow limitation were observed.
Discussion
This study demonstrates that in the bronchial biopsies of smokers with normal lung function and in smokers with mild-to-moderate COPD there is a two to four-fold increase in the expression of p65 protein in comparison with bronchial biopsies from control nonsmokers. Using confocal microscopy nuclear p65 staining is increased in healthy smokers and COPD subjects compared with controls. Furthermore, in subjects with mild-to-moderate COPD, the number of p65+ epithelial cells is increased by 100% and significantly correlated with the degree of airflow limitation. Furthermore, the significant negative correlation between functional parameters (FEV1% and FEV1/FVC%) and numbers of p65+ cells in the epithelium, also suggests a relationship between increasing severity of the disease and epithelial p65 expression in subjects with COPD.
In previous studies performed in patients with mild-to-moderate COPD 4–7 an increased number of T-lymphocytes and macrophages, as well as an increased presence of ICAM-1 in epithelium 5, endothelial leucocyte adhesion molecule-1 in endothelium 5 and macrophage inflammatory protein-1β in the bronchoalveolar lavage fluid 8 have been reported. A number of signs of inflammation, such as MCP-1 overexpression 8 have also been observed in smokers with normal lung function, even though to a lesser extent than in COPD patients 9. However, the role of transcription factors that can modulate the rate of transcription of many inflammatory proteins (IL-1, IL-6, IL-8, MCP-1, TNF-α, ICAM-1) has not been investigated in bronchial biopsies from COPD patients.
As the inflammatory cell infiltration has been reported to be increased during exacerbations of COPD 9, it is speculated that repeated bouts of viral and bacterial origin, may induce a cascade of events resulting in NF-κB induction and activation, cytokine and chemokine production and further inflammatory cell infiltration. Unfortunately, collection of bronchial biopsies in subjects undergoing exacerbations is difficult to perform and rarely accepted by the patients. As such, direct evidence of NF-κB involvement in the bronchial inflammation during exacerbations of COPD is difficult to obtain. However, the increased number of cells expressing nuclear p65 suggests that they may be more activated in response to normal inflammatory insults than cells from normal nonsmoking subjects. This has been reported in many cell types where overexpression of p65 results in stimulation of inflammatory genes 23 and also in cancer-cell malignancy 30. This may help to account for the increasing number and severity of exacerbations seen in these subjects following similar stimuli.
The ability of a cell to activate NF-κB depends upon the relative levels of NF-κB and inhibitor proteins. Under normal resting conditions all NF-κB is retained within the cytoplasm by IκB. Enhanced expression of NF-κB, or the p65 subunit as described here, can overcome the inhibitory effects of IκB thus leading to nuclear translocation and the increased inflammatory gene expression seen in the airways of smokers and subjects with COPD 4–9. This is most obviously seen in in vitro over-expression studies where p65 over-expression negates the requirement for exogenous stimuli to activate NF-κB responsive genes 31. This may be occurring here since smokers and subjects with COPD have increased p65 localisation compared with normal control subjects and this may in turn induce the cytokine production and ICAM-1 over-expression reported in the epithelium of smokers and COPD patients 4–9. The authors believe that the increased p65 expression is not due to increased cellular infiltration, although this does occur, since they express less p65 per cell and are fewer in number than epithelial cells.
The observation that increased p65 expression is present not only in the epithelium of COPD patients but also in the epithelium of smokers with normal lung function suggests that cigarette smoke per se is able to induce this epithelial-cell activation. More than 4,000 particles and oxidants have been described in cigarette smoke 32, which may constitute a continuous external challenge to the bronchial epithelial cells of smoking subjects. This, in turn, may induce NF-κB expression and/or activation in bronchial epithelial cells making them potentially more able to induce pro-inflammatory cytokine production.
As T-lymphocytes represent one of the major sources for cytokine production, in a subset of COPD patients (six subjects) double staining was performed for identification of CD4+ and CD8+ cells that co-expressed p65. It was found that approximately one-third of both T-cell subsets expressed p65. These data suggest that both T-cell subsets may be potentially involved in the cytokine production induced by nuclear expression of p65 in subjects with COPD.
To conclude, it has been shown that the total amount of p65, as assessed by Western blotting, is increased in bronchial biopsies of smokers with normal lung function and in chronic obstructive pulmonary disease patients. This appears to be due to increased numbers of p65+ epithelial cells in comparison to control nonsmokers, as shown by immunohistochemistry. These results indicate a prominent role for epithelial cells in all smokers, as a source of nuclear factor-κB protein. A positive correlation between the degree of airflow limitation and the number of p65+ epithelial cells in subjects with chronic obstructive pulmonary disease is also reported, suggesting that changes in the epithelial p65 expression may be associated with the state of the disease.
- Received August 13, 2001.
- Accepted March 28, 2002.
- © ERS Journals Ltd
References









