





Abstract
Congenital central hypoventilation syndrome is a rare disorder characterised by chronic alveolar hypoventilation, which becomes more pronounced during sleep and may be associated with neurocristopathies, such as Hirchsprung's disease. A mutation in the PHOX2B gene has recently been identified.
In a family of both parents and five offspring, detailed clinical assessment, pulmonary function testing, overnight sleep studies and ventilatory responsiveness to progressive hypercapnia (V′R,CO2) were performed, in addition to analysis of known genetic loci for this condition.
The father and four of the offspring demonstrated features of central hypoventilation with nonapnoeic oxygen desaturation during sleep and diminished V′R,CO2, despite normal pulmonary function. The lowest sleep saturation was median (range) 79% (67–83%) and V′R,CO2 was 2.1 (0.03–4.3) L·min-1·kPa-1. The normal values for the authors’ centre (St Vincent's University Hospital, Dublin, Ireland) are 15–40 L·min-1·kPa-1. An in-frame five amino acid polyalanine expansion of the PHOX2B gene was found in all affected subjects, while the mother and fifth child, who did not have features of central hypoventilation, had a normal PHOX2B gene. Magnetic resonance imaging of the brainstem in one severely affected child was normal.
The present study of a unique family confirms that transmission of late-onset congenital central hypoventilation syndrome is autosomal dominant in nature.
Congenital central hypoventilation syndrome (CCHS), also referred to as Ondine's curse, is a rare condition characterised by ventilatory impairment that results in arterial hypoxaemia, which is worsened by sleep and occurs in patients with normal mechanical properties of the lung. It is diagnosed in the absence of primary neuromuscular, pulmonary or cardiac disease, or an identifiable brainstem lesion 1. The syndrome is usually idiopathic but may occur in association with Hirschprung's disease and disorders of neural crest origin. Studies in monozygotic twins, female siblings, male and female half-siblings, and mother–child transmission support a genetic aetiology 2–6. More recently, an autosomal dominant mode of inheritance has been proposed following the finding of high-frequency (91–97%) heterozygous expansion mutations in a polyalanine tract within exon 3 of PHOX2B gene 7, 8. Although patients typically present in the newborn period and rarely in later infancy, there have been reports of patients presenting with CCHS in adulthood 9–16. Initially, it was thought that PHOX2B mutations occurred de novo 17, 18. However, Weese-Mayer et al. 16 recently described a father and daughter with late-onset CCHS (LOCHS) and the same PHOX2B mutation, indicating autosomal dominant inheritance 16. The present study reports the first complete family in which one parent and four out of five offspring demonstrated features of LOCHS, with genetic analysis confirming the presence of the same PHOX2B expansion mutation.
METHODS
Following identification of central hypoventilation in two siblings, a detailed evaluation was performed of the complete family, consisting of both parents and five offspring. In addition to a full clinical assessment, overnight supervised polysomnography was performed in a sleep laboratory in both parents and the three oldest children using standard techniques 19; home overnight oximetry was performed in the two youngest children. Pulmonary function testing, including spirometry, diffusing capacity and testing of respiratory muscle strength, was performed using a proprietary system (Jaeger, Wurzburg, Germany) and arterial blood gas analysis was performed by radial/femoral arterial puncture. Ventilatory responsiveness to progressive hypercapnia (V′R,CO2) was measured using the Read rebreathing technique in all but the two youngest children 20. Briefly, subjects rebreathed into an airtight 5-L bag containing a mixture of 7% carbon dioxide and 93% oxygen. The spirometer technology used to monitor ventilation was based on a bi-directional rotating vane principle (flow sensing), which was calibrated by a 3-L syringe prior to testing. A continuous record of CO2 concentration in the expired gas was obtained by a CO2 analyser within the circuit. At commencement of the test, subjects took three deep breaths to ensure rapid equilibration between the rebreathing bag, mixed venous CO2 tension (PCO2) and gas in the lung. Rebreathing continued for 4–6 min or until onset of subject discomfort. Discarding the plateau phase of the graph, ventilatory hypercapnic drive was calculated from the slope produced by change in ventilation (L·min-1) and change in end-tidal PCO2.
Genetic studies were performed on venous blood samples to identify a genetic basis for hypoventilation. Linkage studies were performed using polymorphic anonymous markers, closely linked to a series of genes known to be associated with neurocristopathies, and PCR amplification by denaturing high-power liquid chromatography in addition to direct sequencing of PCR product.
RESULTS
Clinical details
Index cases
An 11-yr-old male presented with recurring episodes of hypercapnic respiratory failure. In the previous 5 yrs he had three episodes of pneumonia complicated by hypercapnic respiratory failure, requiring mechanical ventilation on one occasion. He had no clinical features of neuromuscular, cardiac or respiratory disease, or an underlying hypothalamic/endocrine disorder. Overnight polysomnography showed persistent nonapnoeic oxygen desaturation not unusual to rapid eye movement (REM) sleep and occurring with equal frequency in non-REM sleep. Although overnight capnometry was not performed, daytime arterial blood gas analysis showed hypoxaemia and hypercapnia, consistent with a central hypoventilation disorder (table 1⇓) 19. No pathological arrhythmia or evidence of sinus arrest was identified on overnight ECG tracing in any family member. The echocardiograph of the index case was reported as showing a dilated right heart, mild tricuspid regurgitation and an estimated pulmonary artery systolic pressure of 38 mmHg. Chest radiography, pulmonary function testing and magnetic resonance scanning of the brain were normal. LOCHS was diagnosed and the patient was later started on domiciliary nocturnal noninvasive ventilation (NIV).
Pulmonary function and sleep data of the index case’s family
Two years later, the older sister of the index case, then aged 14 yrs, presented with a similar history of recurrent respiratory tract infections complicated by severe hypercapnia requiring invasive ventilatory support. Similar investigations were undertaken, which were in keeping with central hypoventilation (table 1⇑, sibling 2), and she was also started on domiciliary nocturnal NIV.
Family details
The cases of the two siblings prompted the current authors to screen the remaining family for LOCHS, which consisted of the parents (aged 41 and 39 yrs), a son (aged 11 yrs) and two daughters (aged 6 and 4 yrs). All were asymptomatic, had no clinical features of autonomic nervous system dysfunction, no past medical history and had normal pulmonary function tests. Arterial blood gas analysis and overnight sleep studies revealed evidence of nocturnal hypoventilation in the father and four out of the five children (excluding the second youngest) to varying degrees of severity (table 1⇑). V′RCO2 was reduced in the father and three eldest siblings, and showed borderline values in the mother (table 1⇑; fig. 1⇓) 20. In the mother, two overnight polysomnogram studies demonstrated occasional transient, episodic oxygen desaturation into the mid 80% range without associated central or obstructive apnoea (table 1⇑), but the mother did not consent to arterial blood gas analysis. An enquiry regarding the extended family did not identify any other member with a history suggestive of this disorder.

Ventilatory response to progressive hypercapnia in the index case performed by the rebreathing technique. V′E: minute ventilation; PET,CO2: end-tidal carbon dioxide tension.
Follow-up sleep studies after an average of 5 yrs showed deterioration in several family members (table 1⇑). On repeat sleep studies, both the index case and father had an elevated apnoea/hypopnoea index of 17 and 36 episodes·h-1, respectively. However, neither had symptoms of an obstructive sleep apnoea syndrome, such as snoring or excessive daytime sleepiness, and both had an Epworth sleepiness score of one out of a maximum 24 21. Indeed, in this family, quality of life scores, as measured by short form-36, Hospital Anxiety and Depression scores and Epworth scores, did not deviate from normal subjects once matched for age and sex 21–23.
Genetic findings
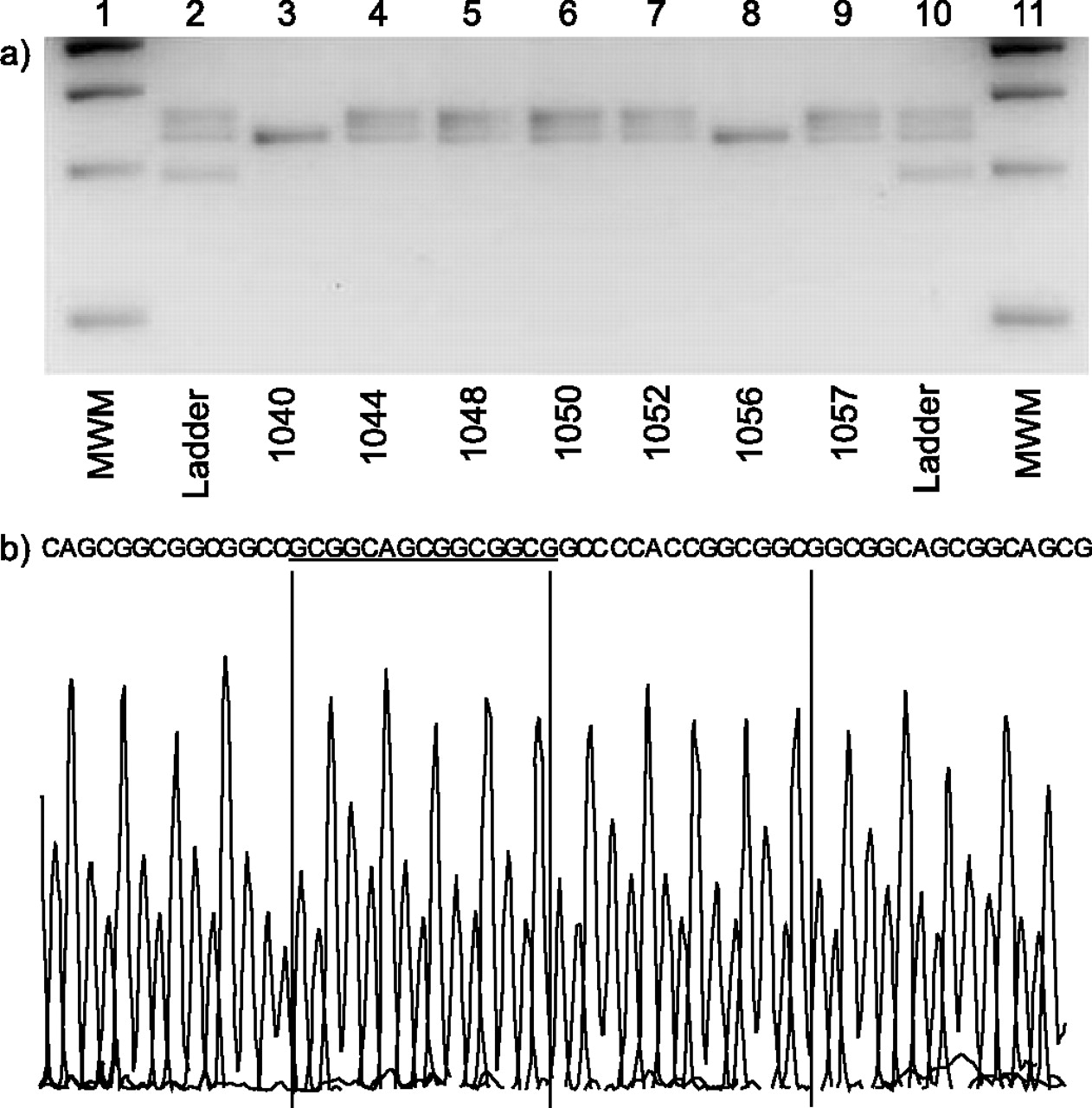
Linkage studies of genes associated with neurocristopathies, namely RET, GDNF, EDN3, EDNRB, BDNF, SOX10, ECE1 and NTN, showed no evidence of association with the disease. An associated gene, hASH1, was also excluded by linkage. In addition, no mutations were found in the hASH1, RET or GDNRF genes using PCR amplification of all exons and direct sequencing of PCR products. However, on amplification and sequencing of the PHOX2B gene, the father and four affected children all demonstrated a 15 base pair insertion at nucleotide 721 of the PHOX2B gene (fig. 2⇓). This in-frame insertion is predicted to give a five amino acid polyalanine expansion previously seen in CCHS, and reported only recently in a delayed onset case of CCHS 16. The mother and unaffected child had a normal PHOX2B gene analysis.
{kind=link}
{kind=link}
a) Polyalanine expansion mutations of the PHOX2B gene in a family with late-onset congenital central hypoventilation syndrome (LOCHS). PCR products of the polyalanine tract (exon 3) from a LOCHS family were analysed by gel electrophoresis on 4% (weight/volume) TBE agarose gels, stained with 0·6 mg·mL-1 ethidium bromide and visualised under ultraviolet light. Lanes 1 and 11: molecular weight marker; lanes 2 and 10: allelic ladder (10/20/25), containing alleles 10Ala (-30 base pairs), normal 20Ala and +5Ala (+15 base pairs); lane 3: mother (20/20), unaffected; lane 4; father (20/25), affected; lane 5: daughter (20/25), affected; lane 6: son (20/25), affected; lane 7: son (20/25), affected; lane 8: daughter (20/20), unaffected; lane 9: daughter (20/25), affected. b) Sequence electropherogram of the +5 polyalanine tract (exon 3) expansion allele indicating the 15 base pair insertion.
DISCUSSION
The present study reports a unique occurrence of a central hypoventilation disorder in most members of a family, which is associated with a known genetic mutation and is in keeping with a diagnosis of LOCHS. However, none of the family members complained of any symptoms of nocturnal hypoventilation or demonstrated any of the neurological, cardiac or ophthalmological features frequently seen in children with CCHS. Had it not been for the screening by the current authors, it is likely the family would have remained undiagnosed 24; however, the current authors recognise that the condition may have been present, but undetected, for many years previously. LOCHS is considered by some as synonymous with a distinct clinical entity of late-onset (childhood) CCHS, in association with hypothalamic dysfunction 25. There are other case reports of adult-onset CCHS in the literature, but the small numbers may reflect the difficulty in recognising a condition that, as in this family, lacks symptoms 9–16. Long-term prognosis remains unclear but the presence of cor pulmonale in two siblings and the interval deterioration sleep-related breathing disturbances are of concern (table 1⇑).
Nocturnal hypoventilation could not be confirmed with overnight capnometry. However, with daytime arterial blood gas analysis in keeping with hypoventilation, normal chest radiographs and lung function tests, and nocturnal hypoxaemia without central or significant obstructive apnoeic episodes on the polysomnogram, the likelihood of an alternative diagnosis is low. Of concern is the elevated body mass index (BMI) in two of the affected siblings (sibling 3 at presentation and sibling 2, whose BMI was 35 kg·m-2 on follow up), since obesity can aggravate nocturnal hypoventilation by obesity displacement of the diaphragm in the supine position. The borderline V′R,CO2values and mild oxygen desaturation in the mother raises the possibility of central hypoventilation, but far less than in the family members showing the PHOX2B mutation. However, the mother’s high BMI also raises the possibility of obesity-related hypoventilation, which is also recognised to result in a low ventilatory drive pattern. 26.
The PHOX2B gene located at chromosome 4p12 encodes a highly conserved homeobox domain transcription factor and was proposed as the disease-defining gene for CCHS by Amiel et al. 17. PHOX2B is a transcriptional activator required for neurogenesis and pan-neuronal differentiation, including type-specific differentiation into the carotid body, the petrosal chemoreceptors that innervate it and the solitary tract nucleus onto which they project 27. Mutations of this gene lead to a variety of disturbances of autonomic function, including CCHS and tumours of the sympathetic nervous system 28.
How LOCHS differs genetically from the usual early presentation of CCHS is unclear and may simply reflect a separate polymorphism or a hitherto unknown gene regulating the activity or expression of PHOX2B. PHOX2B polyalanine repeat expansion mutation length also correlates with clinical features of autonomic nervous system dysfunction in CCHS patients 8. To date, only 14 patients have been reported who have LOCHS and were tested for the PHOX2B mutation 7, 16, 28, 29. Among them, 10 patients carried a PHOX2B mutation with +5 alanine expansion, which is similar to the family members carrying a PHOX2B mutation in the present study.
The present findings confirm that the small alanine expansion (+5) is present in late-onset congenital central hypoventilation syndrome patients, compared with the longer expansion reported in patients with congenital central hypoventilation syndrome or congenital central hypoventilation syndrome plus Hirschsprung's disease. The current study represents the only complete family study of this mutation and the findings indicate an autosomal dominant mode of inheritance.
Acknowledgments
The authors are grateful to D. Gill (Temple Street Children's Hospital, Dublin, Ireland) for referring the index case and child 2. The authors are also grateful to the staff of the Pulmonary Function and Sleep Laboratories (St. Vincent's University Hospital, Dublin) for performing pulmonary function and sleep studies.
- Received January 5, 2006.
- Accepted September 21, 2006.
- © ERS Journals Ltd
References










