





Abstract
Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated nuclear hormone receptors belonging to the steroid receptor superfamily. Previously, the present authors have shown that PPAR‐γ agonists inhibit the release of inflammatory cell survival factors and induce apoptosis in vitro. The aim of this study was to determine the effect of two structurally different PPAR agonists in an in vivo model of lipopolysaccharide (LPS)-induced airway inflammation.
Mice were treated with PPAR agonists, rosiglitazone or SB 219994, prior to exposure to aerosolised LPS, and the extent of airway inflammation was assessed 3 h later.
In these experiments, the PPAR ligands inhibited LPS-induced airway neutrophilia and associated chemoattractants/survival factors (keratinocyte-derived chemokine and granulocyte-colony stimulating factor) in the mouse lung.
The present authors postulate that if a peroxisome proliferator-activated receptor agonist has the same effect in man, and neutrophils are important in the progression of respiratory diseases, such as chronic obstructive pulmonary disease, then this class of compounds could be a potential therapy. Furthermore, several peroxisome proliferator-activated receptor‐γ agonists have been shown to be clinically effective for the treatment of type II diabetes, suggesting that any benefit of peroxisome proliferator-activated receptor‐γ ligands in the progression of respiratory diseases, which may involve airway neutrophilia, could be explored relatively quickly.
This work was supported by the Harefield Research Foundation and a grant from the Clinical Research Committee, Royal Brompton and Harefield Hospital Trust.
Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated nuclear hormone receptors belonging to the steroid receptor superfamily 1. Three different PPAR subtypes have been identified to date, including PPAR‐α, ‐γ and ‐δ (also referred to as nuclease abnormal‐1 (NUC‐1), PPAR‐β or fatty acid activated receptor (FAAR)). PPARs were first identified for their role in lipid and glucose regulation (energy balance), and until recently their actions were thought to be limited to specific tissue types. PPAR‐α ishighly expressed in tissues exhibiting high carbolic rates offatty acids, such as the liver, heart, kidney and intestinal mucosa, and has also been shown to be present in the lung 2–4. PPAR‐γ, as well as being highly expressed in adipose tissue, where it plays a critical role in adipocyte differentiation 5, has been found in a number of cell types, including airway smooth muscle, epithelial cells and macrophages 6, 7. PPAR‐δ is almost ubiquitously expressed 2, 3, 8, however, its role is relatively unknown. Recently, it has been suggested that PPAR‐α and PPAR‐γ are important immunomodulators 9. PPAR‐α activation inhibits inflammatory mediator release from vascular smooth muscle cells 10 and is consistent with the finding that PPAR‐α knockout mice exhibit exacerbated inflammatory responses 11. PPAR‐γ ligands have been shown to inhibit the release of pro-inflammatory cytokines from activated macrophages 12 and airway epithelial cells 13. Furthermore, PPAR‐γ ligands have been shown to inhibit vascular smooth muscle cell proliferation 14, and induce apoptosis in endothelial cells 15, vascular smooth muscle cells 16, T‐lymphocytes 17, 18 and macrophages 19.
The current authors have previously reported that activation of PPAR‐γ has potent anti-inflammatory effects on human airway smooth muscle (HASM) cells in culture. In particular, PPAR‐γ activation inhibits the release of survival factors and smooth muscle cell proliferation, as well as inducing apoptosis. Previously in these studies, the current authors have shown that PPAR‐γ activation has a superior anti-inflammatory profile to that of a glucocorticoid, dexamethasone, with respect to its effects on inflammatory mediator release from this cell type 6. Hence, the aim was to further investigate the anti-inflammatory effect of PPAR‐γ activation in an in vivo model of airway inflammation. Two structurally distinct PPAR agonists, rosiglitazone maleate and SB 219994, were used in an attempt to demonstrate that any effect is related to the activation of the receptor, rather than through nonspecific actions of the compound. Airway inflammation was triggered by exposure to aerosolised LPS, which is known to induce neutrophilia and associated chemoattractants/survival factors such as keratinocyte-derived chemokine (KC), granulocyte-colony stimulating factor (G‐CSF) and granulocyte-macrophage-colony stimulating factor (GM‐CSF) in the airways. Treatment with dexamethasone was included as a positive control for the experiments.
Methods
Animals
Male BALB/c mice (14–16 g) were obtained (Harlan, Bicester, Oxon, UK) and housed for 1 week before initiating experiments. Food and water were supplied ad libitum. Experiments were performed in accordance with the UK Home Office guidelines for animal welfare based on the Animals (Scientific Procedures) act 1986.
Effect of compounds on LPS-induced increase in cytokine levels in the mouse lung
Male BALB/c mice were orally dosed with vehicle (methylcellulose, 0.5% and tween 80 0.2%, 10 mL·kg−1), rosiglitazone maleate (1, 3, 10, 30 or 100 mg·kg−1) (GlaxoSmithKline, UK), SB 219994 (1, 3, 10, 30 or 100 mg·kg−1) (GlaxoSmithKline) or dexamethasone (1 mg·kg−1) 1 h prior to challenge with aerosolised saline or LPS (0.1 mg·mL−1 for 30 min). The mice were sacrificed with an overdose of pentobarbitone (200 mg·kg−1 i.p.) 3 h after challenge, the lungs were lavaged with RPMI (Invitrogen, Paisley, UK) (three 0.3 mL washes, pooled to give a total recovered volume of 0.5–0.6 mL) and then removed, flash frozen in liquid nitrogen and stored at −80°C until required.
The lung tissue was homogenised in 2 mL of ice-cold saline and centrifuged at 800×g. The resulting supernatant and lavage fluid was assessed for G‐CSF, GM‐CSF and KC content by ELISA (R&D systems, Abington, UK) in accordance with manufacturer's instructions and expressed per mL or per mg of protein. All other materials were purchased from Sigma-Aldrich Chemical Company, Poole, UK.
Effect of compounds on LPS-induced airway neutrophilia in the mouse
Male BALB/c mice were orally dosed with vehicle (methylcellulose, 0.5% and tween 80 0.2%, 10 mL·kg−1), rosiglitazone maleate (30 mg·kg−1), SB 219994 (30 mg·kg−1) or dexamethasone (1 mg·kg−1) 1 h prior to challenge with aerosolised saline or LPS (0.1 mg·mL−1 for 30 min). The mice were sacrificed with an overdose of pentobarbitone (200 mg·kg−1 i.p.) 3 h after challenge, the lungs were lavaged with RPMI (three 0.3 mL washes, pooled) and then removed, perfused to remove the blood pool and finely chopped. The inflammatory cells were extracted from the lung tissue by collagenase digest as described by Underwood et al. 20. Collagenase and DNase were purchased from Roche Diagnostics (Lewes, UK). Total white cell number in the lavage fluid and lung tissue preparations was determined on a Sysmex F820 (Linford Wood, Milton Keynes, UK). Differential white cellcounts were determined using standard morphological criteria.
Effect of compounds on LPS-induced increases in MMP‐9 levels in the mouse lung
Lavage and tissue homogenate supernatants (from the study above) were assessed for MMP‐9 content by zymography in accordance with manufacturer's instructions (Invitrogen). Briefly, supernatants were denatured, run on gels containing gelatin, re-natured and left over night at 37°C. The gels were then stained for protein and the absence of colour indicated the presence of MMP. The amount of MMP present was semiquantitated on a UVP Bio-imaging system.
Effect of rosiglitazone, when dosed after insult, on LPS-induced airway inflammation
Male BALB/c mice were orally dosed with vehicle (methylcellulose, 0.5% and tween 80 0.2%, 10 mL·kg−1), rosiglitazone maleate (30 mg·kg−1), or dexamethasone (1 mg·kg−1) 1 h after challenge with aerosolised LPS (0.1 mg·mL−1 for 30 min). The mice were sacrificed with an overdose of pentobarbitone (200 mg·kg−1 i.p.) 3 h after challenge. Levels of KC, G-CSF, GM-CSF and numbers of neutrophils were assessed in the lung tissue according to the methods described above.
Statistical analysis
Data are presented as the mean±sem per group, n=8. Statistical analysis was made for multiple comparisons using analysis of variance with an appropriate post hoc test. A p‐value of <0.05 was considered to be statistically significant.
Results
Effect of compounds on LPS-induced increase in cytokine levels in the mouse lung
Aerosolised LPS challenge evoked a significant increase in G‐CSF, GM‐CSF and KC levels in the lavage fluid and lung tissue (figs 1⇓ and 2⇓). Treatment with either PPAR agonist reduced the LPS-induced rise in G‐CSF and KC levels in the lung tissue but not in the lung lavage fluid. Neither PPAR ligand had any impact on LPS-induced increases in GM‐CSF levels in the lung tissue or lavage fluid. Neither agonist had any effect on nonstimulated levels of any of the cytokines measured. The positive standard, dexamethasone, significantly inhibited all three cytokine levels in the lung tissue but only GM‐CSF levels in the lavage fluid (figs 1⇓ and 2⇓).

Effect of compounds on lipopolysaccharide (LPS)-induced increases in granulocyte-colony stimulating factor (G‐CSF), granulocyte-macrophage colony stimulating factor (GM-CSF) and keratinocyte-derived chemokine (KC) levels in the mouse lung lumen. Mice were orally dosed with vehicle (□), rosiglitazone (▪), SB 219994 ( ) or dexamethasone (
) or dexamethasone ( ) 1 h prior to challenge with aerosolised saline or LPS. Supernatant from the lung lavage was assessed for a) G‐CSF, b) GM‐CSF and c) KC content by ELISA. Data are expressed as mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
) 1 h prior to challenge with aerosolised saline or LPS. Supernatant from the lung lavage was assessed for a) G‐CSF, b) GM‐CSF and c) KC content by ELISA. Data are expressed as mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.

Effect of compounds on lipopolysaccharide (LPS)-induced increases in granulocyte-colony stimulating factor (G‐CSF), granulocyte-macrophage colony stimulating factor (GM‐CSF) and keratinocyte-derived chemokine (KC) levels in the mouse lung tissue. Mice were orally dosed with vehicle (□), rosiglitazone (▪), SB 219994 ( ) or dexamethasone (
) or dexamethasone ( ) 1 h prior to challenge with aerosolised saline or LPS. Supernatant from the lung tissue homogenate was assessed for a) G‐CSF, b) GM‐CSF and c) KC content by ELISA. Data are expressed as the mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
) 1 h prior to challenge with aerosolised saline or LPS. Supernatant from the lung tissue homogenate was assessed for a) G‐CSF, b) GM‐CSF and c) KC content by ELISA. Data are expressed as the mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
From these experiments it was determined that a dose of 30 mg·kg−1 of both PPAR agonists was sufficient to inhibit LPS‐induced cytokine release and this dose was used in the next experiment examining the effect of these compounds on cell recruitment.
Effect of compounds on LPS‐induced airway neutrophilia in the mouse
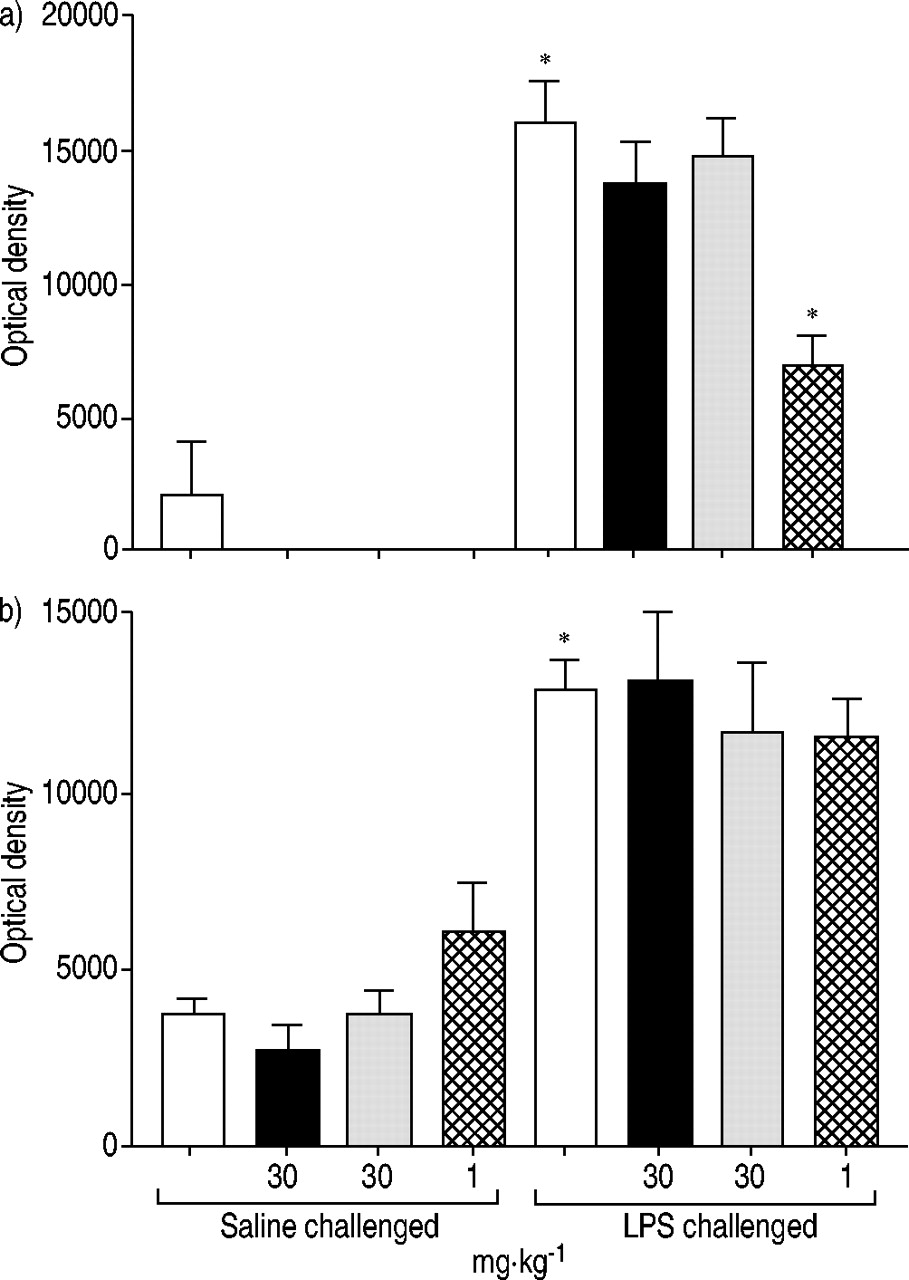
Aerosolised LPS exposure caused a significant increase in both lung lavage and tissue numbers of neutrophils (fig. 3⇓). Neither ligand had any effect on cell number in the lavage fluid, whereas dexamethasone significantly inhibited neutrophil number. Rosiglitazone, SB 219994 and dexamethasone blocked lung tissue neutrophilia (49.7, 33.1 and 58.2% inhibition, respectively), with the effects of rosiglitazone and dexamethasone reaching statistical significance and being of similar magnitude (fig. 3⇓). None of the PPAR ligands had any significant effect on nonstimulated neutrophil number in the airways, whereas dexamethasone significantly reduced basal lung tissue levels.

Effect of compounds on lipopolysaccharide (LPS)-induced airway neutrophilia in the mouse. Mice were orally dosed with vehicle (□), rosiglitazone (▪), SB 219994 ( ) or dexamethasone (
) or dexamethasone ( ) 1 h prior to challenge with aerosolised saline or LPS. Lung a) lumen and b) tissue inflammatory cell content were assessed. Data are expressed as mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
) 1 h prior to challenge with aerosolised saline or LPS. Lung a) lumen and b) tissue inflammatory cell content were assessed. Data are expressed as mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
LPS exposure did not change the numbers of lymphomononuclear (LMN) cells in BAL fluid and lung tissue. Neither PPAR‐γ ligand altered the numbers of LMN in the BAL (47±15, 50±10, 62±17 cells×103·mL−1 for vehicle, rosiglitazone and SB 219994, respectively) or lung tissue (5590±771, 4137±547, 492837±589 cells×103·mg−1 tissue for vehicle, rosiglitazone or SB 219994, respectively). The positive standard, dexamethasone, did not affect BAL fluid LMN number (55±4 cells×103·mL−1) but significantly decreased lung tissue LMN numbers (2329±359 cells×103·mg−1).
Effect of compounds on LPS-induced increase in MMP‐9 levels in the mouse lung
Neither ligand had any effect on lavage or lung tissue homogenate MMP‐9 content at the doses tested, whereas dexamethasone significantly inhibited MMP‐9 levels in the lavage fluid, as measured by zymography (fig. 4⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of compounds on LPS-induced increases in matrix metalloproteinase‐9 (MMP‐9) levels in the mouse lung. Mice were orally dosed with vehicle (□), rosiglitazone (▪), SB 219994 ( ) or dexamethasone (
) or dexamethasone ( ) 1 h prior to challenge with aerosolised saline or LPS. Lung a) bronchoalveolar lavage and b) tissue homogenate supernatants were assessed for MMP‐9 content by zymography. Data are expressed as mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
) 1 h prior to challenge with aerosolised saline or LPS. Lung a) bronchoalveolar lavage and b) tissue homogenate supernatants were assessed for MMP‐9 content by zymography. Data are expressed as mean±sem. *: p<0.05 compared with the relevant vehicle control group. n=8.
Effect of rosiglitazone, when dosed after insult, on LPS-induced airway inflammation
Rosiglitazone, when dosed after the LPS insult, caused a 41% reduction in lung tissue G‐CSF levels and a 26% reduction in lung tissue neutrophilia (table 1⇓) compared with the vehicle-treated group. The PPAR‐γ ligand failed to impact on KC or GM‐CSF levels when dosed after the LPS challenge, whereas dexamethasone caused a significant reduction in allthree cytokines but, interestingly, not on lung tissue neutrophilia (table 1⇓).
Effect of rosiglitazone on lung tissue inflammation, administered after the lipopolysaccharide (LPS) insult
Discussion
This study demonstrates that PPAR agonists inhibit LPS‐induced release of pro-inflammatory cytokines, G‐CSF and KC, which was associated with a reduction in lung tissue neutrophil number. This effect was seen with two structurally distinct molecules suggesting the response was due to PPAR agonist activity, and the effect of rosiglitazone was of a similar magnitude to that of the steroid, dexamethasone. It appears from the magnitude of impact of the two PPAR ligands on lung tissue neutrophil number that rosiglitazone is more effective than SB 219994. This is surprising as SB 219994 is reported to be more potent than rosiglitazone both in vitro and in vivo 21. A possible reason for this is the fact that, unlike rosiglitazone which is a selective PPAR‐γ agonist, SB 219994 has recently been shown to be a dual PPAR‐α/γ agonist 22. Although the agonists reduced lung tissue neutrophil number they did not alter lung lavage numbers. This lack of effect in the airway lumen concurs with a recent publication by Trifilieff et al. 23 in which they reported that PPAR ligands did not inhibit LPS-induced increases in lung lavage neutrophil number. The reason why the PPAR ligands only affected lung tissue is not known. However, it could be argued that inhibition of cell infiltration into the lung tissue is the more relevant compartment to achieve efficacy with drug therapy. Interestingly, the impact of the PPAR ligands on cytokine release in the model appears to be selective for the lung tissue compartment, and not the airway lumen, and for G‐CSF and KC and not GM‐CSF or MMP‐9. The reasons for these selective effects are not clear and contradict some of the reported anti-inflammatory activities of PPAR ligands seen in vitro on GM‐CSF and MMP‐9 release 6, 24. However, given that the positive standard, dexamethasone, did not reduce G‐CSF and KC levels in the lavage fluid suggests that the negative data with the PPAR ligands should not be over interpreted.
In an attempt to determine the impact of a PPAR‐γ ligand in a clinical setting, i.e. the impact of the compound once the inflammatory process has been initiated; rosiglitazone (the most effective PPAR‐γ ligand tested previously) or dexamethasone was administered 1 h after the LPS insult. The profile of the results seen using this protocol is similar to that achieved when the compound was administered prior to LPS. Rosiglitazone appeared to selectively inhibit G‐CSF production and neutrophil recruitment. Interestingly, although the steroid inhibited G‐CSF, GM‐CSF and KC levels in the lung, it failed to decrease neutrophil numbers. This may suggest that a PPAR‐γ ligand has additional anti-inflammatory activities to that of a steroid when dosed using this protocol.
Even though the exact mechanism of action of the PPAR‐γ ligands is not known, it appears from this study that they may be of benefit in the treatment of respiratory diseases that are thought to involve airway neutrophilia, such as acute respiratory distress syndrome (ARDS) and chronic obstructive pulmonary disease (COPD). Exactly how the PPAR‐γ ligands are having their impact on this model is not known, however, there are a number of possibilities. First, PPAR‐γ exists inactivated as a heterodimer, with the retinoid X receptor, a member of the orphan nuclear receptor superfamily. After binding of the ligand the corepressors are released and the receptor complex can activate specific genes. These genes may code for anti-inflammatory proteins. An example of this has been reported by Meier et al. 25 in which they demonstrate an upregulation of the anti-inflammatory IL‐1 receptor antagonist (IL‐1Ra) gene expression by PPAR agonists in vitro. Secondly, stimuli such as LPS cause the degradation of inhibitory κB (IκB), allowing nuclear factor-κB (NF‐κB) to migrate to the nucleus where it binds to the promoter of pro-inflammatory genes, such as inducible nitric oxide synthase, cyclooxygenase‐2 and tumour necrosis factor‐α 26. PPAR‐γ ligands have been shown to stabilise the IκB/NF‐κB interaction, blocking NF‐κB entry to the nucleus, thus inhibiting pro-inflammatory gene expression. Thirdly, both the NF-κB and the PPAR‐γ transcription factors utilise many of the same co-activators, thus activation of PPAR‐γ may block activation of NF‐κB by competing for the cofactors 27. Finally, the ligands could be reducing neutrophil number by inducing apoptosis either directly or by interfering with the anti-apoptotic NF‐κB signalling pathway, or by reducing anti-apoptotic cytokines, such as G‐CSF.
In conclusion, the present study found that treatment with rosiglitazone or SB 219994 inhibited lipopolysaccharide-induced neutrophilia and associated chemoattractants/survival factors (keratinocyte-derived chemokine and granulocyte-colony stimulating factor) in the mouse lung. The current authors postulate that if a peroxisome proliferator-activated receptor‐γ agonist has the same effect in man, this class of molecule may have therapeutic potential in respiratory diseases that are thought to involve neutrophilia. Furthermore, several peroxisome proliferator-activated receptors‐γ agonists have been shown to be clinically effective for the treatment of type II diabetes, with minor adverse side-effects, suggesting that any benefit of peroxisome proliferator-activated receptors‐γ ligands in the progression of respiratory diseases, which may involve airway neutrophilia, could be explored relatively quickly.
- Received August 28, 2003.
- Accepted March 14, 2004.
- © ERS Journals Ltd
References









