





Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive disease of the lung parenchyma, causing significant morbidity through worsening dyspnoea and overall functional decline. IPF is characterised by apoptosis-resistant myofibroblasts, which are a major source for the excessive production of extracellular matrix (ECM) overtaking normal lung tissue. We sought to study the role of heat shock protein (HSP) isoforms HSP90α and HSP90β, whose distinct roles in lung fibrogenesis remain elusive.
We determined the level of circulating HSP90α in IPF patients (n=31) and age-matched healthy controls (n=9) by ELISA. The release of HSP90α and HSP90β was evaluated in vitro in primary IPF and control lung fibroblasts and ex vivo after mechanical stretch on fibrotic lung slices from rats receiving adenovector-mediated transforming growth factor-β1.
We demonstrate that circulating HSP90α is upregulated in IPF patients in correlation with disease severity. The release of HSP90α is enhanced by the increase in mechanical stress of the fibrotic ECM. This increase in extracellular HSP90α signals through low-density lipoprotein receptor-related protein 1 (LRP1) to promote myofibroblast differentiation and persistence. In parallel, we demonstrate that the intracellular form of HSP90β stabilises LRP1, thus amplifying HSP90α extracellular action.
We believe that the specific inhibition of extracellular HSP90α is a promising therapeutic strategy to reduce pro-fibrotic signalling in IPF.
Abstract
Extracellular HSP90α and intracellular HSP90β act together to promote lung fibrosis progression http://ow.ly/bAmH30gGQf1
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive disease of the lung parenchyma, causing significant morbidity through worsening dyspnoea, increasing cough and overall functional decline. The precise cause of IPF remains elusive and treatment options are limited, with only two approved drugs for IPF, i.e. nintedanib and pirfenidone, which were shown to slow but not stop disease progression [1, 2]. Epidemiological studies demonstrated that the incidence of IPF has been rising over the past decade, particularly in the age group >70 years [3, 4]. The prognosis of IPF is poor, with most patients succumbing to their illness at a rate comparable to aggressive cancers [5]. IPF is characterised by an increase in apoptosis-resistant myofibroblasts in the lungs, which are a major source for the aberrant deposition of extracellular matrix (ECM), synthesis of pro-fibrotic cytokines such as transforming growth factor (TGF)-β1 and are also associated with disease progression [6]. Fibrotic ECM is stiffer than nonfibrotic ECM, increasing the mechanical stress within the fibrotic lungs [7]. In addition to the biochemical, structural and mechanical features of the ECM, the matrix is also a major storage compartment for growth factors, cytokines and various extracellular proteins that affect the behaviour of surrounding cells. TGF-β1 activation by mechanical stretch plays a crucial role in IPF by inducing myofibroblast differentiation, thus increasing ECM deposition in the fibrotic lung [8]. Myofibroblast differentiation is a central event in fibrogenesis. Resident fibroblasts and epithelial cells, in a process called epithelial-to-mesenchymal transition (EMT), were demonstrated to differentiate into myofibroblasts under TGF-β1 stimulation [9].
HSP90 belongs to the heat shock protein (HSP) family of stress-inducible proteins. Despite their cytoprotective role, HSPs are involved in pathological processes such as cancer progression, metastasis formation and fibrosis development [10]. Two isoforms, HSP90α and HSP90β, that share 86% amino acids are expressed in all nucleated cells. Recently, HSP90α, and to a lesser extent HSP90β, has been shown to be secreted by cancer cells and cells subjected to various stress conditions such as hypoxia, DNA damage and also growth factor stimulation (e.g. TGF-α) [11, 12]. Extracellular HSP90 has been reported to promote cell motility of dermal fibroblasts, accelerating murine skin wound closure and re-epithelialisation in vivo [13]. It has also been shown to be a major regulator of EMT, promoting metastasis formation in a prostate cancer model [14]. It has been demonstrated that extracellular HSP90 activity is driven by low-density lipoprotein receptor-related protein 1 (LRP1) further activating extracellular signal-regulated kinase (ERK) and Akt phosphorylation, and leading to EMT, increasing cell motility and myofibroblast differentiation [15, 16]. However, the distinct roles of the extracellular forms of both HSP90α and HSP90β remain elusive. Here, we demonstrate that circulating HSP90α, but not HSP90β, is upregulated in IPF patients in correlation with disease severity. The release of HSP90α is enhanced by the increase in mechanical stress induced in the fibrotic ECM. This increase in extracellular HSP90α activates, through the receptor LRP1, pro-fibrotic pathways in pulmonary epithelial cells and fibroblasts, and promotes matrix remodelling leading to the establishment of a pro-fibrotic microenvironment allowing myofibroblast differentiation and persistence. In parallel, the intracellular form of HSP90β stabilises LRP1, thus amplifying HSP90α extracellular action. Therefore, we believe that the specific inhibition of extracellular HSP90α is a promising therapeutic strategy to reduce pro-fibrotic signalling in IPF.
Materials and methods
See the supplementary material for detailed methods.
Human samples
All plasma and tissue were collected with patient approval by the Hamilton Integrated Research Ethics Board (00-1839). Control lung tissue was collected from patients undergoing surgery for cancer. Lung fibrosis tissue was collected from patients undergoing biopsy for the diagnosis of unclear interstitial lung disease. The biopsies analysed in this study revealed a usual interstitial pneumonia (UIP) pattern on histopathology.
Animal procedure
Sprague Dawley rats (8 weeks old) were treated according to the guidelines from the Canadian Council on Animal Care and approved by the Animal Research Ethics Board of McMaster University (protocol 13.12.48). Rats received 5.0×108 PFU of adenovector-mediated TGF-β1 (AdTGF-β1) (or control AdDL) by intratracheal instillation under isoflurane anaesthesia as described previously (see supplementary methods for details) [17].
Collagen quantification
Hydroxyproline content in rat lung samples was measured by a colorimetric assay described previously (see supplementary methods for details) [18].
Cell culture
Control and IPF primary lung fibroblasts were isolated as described previously from tumour-free lung areas of three patients undergoing surgery for cancer and from three patients with interstitial lung disease undergoing surgery and eventually being confirmed as having IPF-UIP [19]. Primary lung fibroblasts were cultured in DMEM/10% fetal bovine serum (FBS). A549 cells (ATCC, Teddington, UK) were cultured in DMEM/10% FBS. Cells were cultured on regular tissue culture plates (Thermo Fisher Scientific, Burlington, ON, Canada) or on tissue culture plates coated with hydrogels of various stiffness (Matrigen, Brea, CA, USA; 1 kPa (soft) or 50 kPa (stiff)). When indicated, human primary lung fibroblasts (controls and IPF) were grown on regular tissue culture plates (to allow expansion) until passage 2 and were then cultured on the stiff or soft hydrogel until passage 3 when they were used for the experiments. When indicated, cells were treated with 5 µM (cell treatment) or 10 µM (co-immunoprecipitation) of HS-30, a HSP90 inhibitor that has been specifically designed to be cell impermeable. HS-30 is an HSP90 inhibitor derivative designed as described previously and was kindly provided by Timothy Haystead (Dept of Pharmacology and Cancer Biology, Duke University, Durham, NC, USA) [20, 21]. MG132 (Sigma Aldrich, Oakville, ON, Canada) inhibits proteasome activity and was used 6 h prior to cell harvesting at a concentration of 50 µM. Specific inhibition of HSP90α, HSP90β and LRP1 was achieved using small interfering (si) RNA (HSP90α: s6994; HSP90β: s14375; LRP1: s8279; Thermo Fisher Scientific). Recombinant (r) HSP90α (Enzo Life Science, Farmingdale, NY, USA) was used to treat A549 cells and primary fibroblasts at a concentration of 10 µM for 24–48 h. rTGF-β1 (R&D Systems, Minneapolis, MN, USA) was used to treat A549 cells for 48 h at 2 ng·mL−1. Blocking HSP90α antibody (AbHSP90α) was used to treat A549 cells and primary fibroblasts at a concentration of 40 µM for 24–48 h. SD-208, an inhibitor of TGF-β receptor I (TGF-βRI), was used to treat primary fibroblasts at a concentration of 30 µM for 24–48 h.
Western blotting
Western blotting assay was used to detect HSP90α (ADI-SPS-771-F; Enzo Life Science), HSP90β (ADI-SPA-844-200; Enzo Life Science), TGF-β1 (ab92486; Abcam, Toronto, ON, Canada), α-smooth muscle actin (α-SMA) (ab7817; Abcam), proliferating cell nuclear antigen (ab18197; Abcam), phosphorylated Smad2 (p-Smad2) (#3101; Cell Signaling Technology, Beverly, MA, USA), Smad2 (ab63576; Abcam), collagen 1A1 (ab34710; Abcam), ERK (#9102; Cell Signaling Technology), phosphorylated ERK (p-ERK) (#4377; Cell Signaling Technology), LRP1 (ab92544; Abcam), Na,K-ATPase (#3010; Cell Signaling Technology), TGF-βRI (ab31013; Abcam) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (ab9485; Abcam). See supplementary methods for details.
Immunofluorescence
Immunostaining of HSP90α (ab59459; Abcam) and LRP1 (ab92544; Abcam) was performed on formalin-fixed human lung tissue sections (see supplementary methods for details).
Ex vivo mechanical stretch procedure
Rat fibrotic lung slices underwent our mechanical stretch protocol as described previously (supplementary figures S1) [22]. Samples from the tissue bath taken before the strips had undergone stretch were termed “before”. Between 10 and 15 s of static 5 mN was applied in order to obtain the Young's modulus measurements. Following these measurements, 4 min of 15 mN cyclical stretch was administered and samples from the tissue bath solution samples were collected (termed “after”). Nonstretched lung tissues were termed “unstimulated” and stretched lung tissues were termed “stimulated”.
We aimed to model the tension of breathing in the tissue bath by setting the tension of cyclic stretch to 15 mN for 4 min oscillating at a frequency of 2 Hz, to a length of 1.1 times the original resting length of the tissue strips (lung strips measured 10×2×2 mm). The estimated pressure was 15 mN per 4 mm2=0.37 mN·mm–2=37.73 cmH2O, which is within the pressure range observed in the lungs of patients with IPF [23]. In some experiments lung slices were pre-treated for 2 h with a protein transport inhibitor (eBioscience, San Diego, CA, USA). The stiffness of tissue was expressed as Young's modulus (N·mm–2) as described previously [22]. See supplementary methods for details.
Ex vivo inhibition of extracellular HSP90
Rat fibrotic lung slices were collected from rats receiving AdTGF-β1 at day 21. Lung slices were cultured in DMEM/10% FBS and received HS-30 (10 µM) or vehicle for 72 h. Lung slices were then harvested for mRNA and protein expression analysis.
Co-immunoprecipitation (membrane fractionation)
For LRP1 co-immunoprecipitation, plasma membrane proteins were extracted using a kit (ab65400; Abcam) following the manufacturer's recommendations. Co-immunoprecipitation experiments were performed on plasma membrane fractions or whole-cell extracts using the ExactaCruz kit (Santa Cruz Biotechnology, Dallas, TX, USA) following the manufacturer's recommendations. See supplementary methods for details.
Statistical analysis
All data are presented as mean±sem. Statistical analysis between two groups was performed using a parametric t-test. Statistical analysis between multiple groups with one control group was performed by one-way ANOVA, with Tukey's multiple comparison test (post hoc). Analysis was performed with Prism version 6.0 (GraphPad, La Jolla, CA, USA). A p-value <0.05 was considered significant.
Results
Circulating HSP90α is upregulated in IPF patients and in AdTGF-β1-induced lung fibrosis
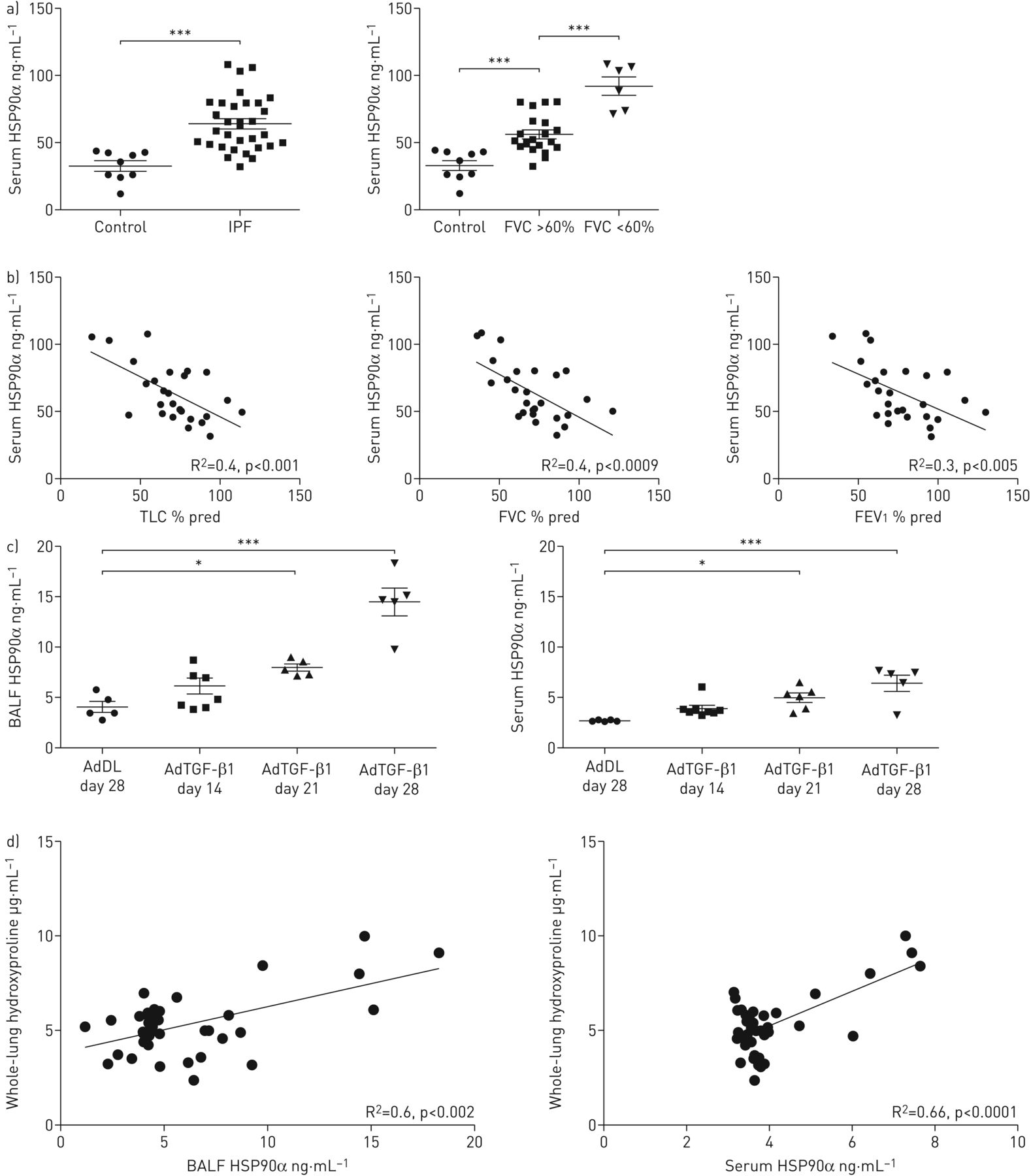
The levels of circulating HSP90α and HSP90β were measured in sera of patients with IPF (n=31) and aged-matched controls (n=9). HSP90α was significantly upregulated in IPF patients compared with controls, whereas HSP90β remained unchanged (figure 1a and supplementary figure S2). To further characterise HSP90α upregulation in IPF, patients were separated into two groups according to their forced vital capacity (FVC) % pred: moderate IPF (FVC % pred >60%, n=25) and severe IPF (FVC % pred <60%, n=6) according to the literature [24]. Interestingly, circulating HSP90α was upregulated in moderate and severe IPF patients compared with controls; severe IPF patients had a significantly higher level of circulating HSP90α compared with moderate IPF patients (figure 1a). HSP90β was not different between groups (supplementary figure S2). Furthermore, the serum level of HSP90α in IPF patients was negatively correlated with lung function tests: total lung capacity, FVC and forced expiratory volume in 1 s (figure 1b).

Circulating heat shock protein (HSP) isoform HSP90α is upregulated in idiopathic pulmonary fibrosis (IPF) patients and in adenovector-mediated transforming growth factor-β1 (AdTGF-β1)-induced lung fibrosis. a) Serum level of HSP90α measured by ELISA on IPF patients and respective aged-matched healthy volunteers. Data presented as mean±sem; n=9 and n=31 for controls and IPF patients, respectively. IPF patients were clustered in two groups: moderate IPF (FVC % pred >60%) and severe IPF (FVC % pred <60%). Data presented as mean±sem; n=9, n=25 and n=6 for controls, moderate IPF and severe IPF patients, respectively. b) Correlation curves between the serum level of HSP90α in IPF patients and clinical lung function parameters: total lung capacity (TLC), forced vital capacity (FVC) and forced expiratory volume in 1 s (FEV1). c) Bronchoalveolar lavage fluid (BALF) and serum levels of HSP90α of rats treated with AdTGF-β1 (or AdDL as control). Data presented as mean±sem; AdDL: n=5; AdTGF-β1 day 14: n=8; AdTGF-β1 day 21: n=5; AdTGF-β1 day 28: n=5. d) Correlation curves between BALF and serum levels of HSP90α and hydroxyproline lung content in rats treated with AdTGF-β1. *: p<0.05; ***: p<0.001.
Rats receiving AdTGF-β1 intratracheally develop a strong progressive fibrosis by day 28 after injection [17]. Western blot analysis revealed that the HSP90α level was upregulated in bronchoalveolar lavage fluid (BALF) from fibrotic rats treated with AdTGF-β1 at day 28 (supplementary figure S2). This result was confirmed by ELISA in BALF and sera of AdTGF-β1-treated rats (figure 1c). Interestingly, the extracellular level of HSP90α gradually increased from day 14 (early fibrosis) up to day 28 (late established fibrosis), suggesting a correlation between HSP90α level and fibrosis progression. Indeed, the extracellular level of HSP90α in BALF and sera of AdTGF-β1-treated rats was correlated with the amount of hydroxyproline, an indirect quantification of the increase in collagen occurring during fibrosis, and the fibrosis score (Ashcroft score) of the lungs (figure 1d and supplementary figure S2).
Taken together, these results demonstrate that circulating HSP90α, but not HSP90β, is upregulated in IPF patients and in rats with pulmonary fibrosis in correlation with fibrosis severity.
Mechanical stretch and tissue stiffness induce HSP90α secretion
The secretion of HSP90α and HSP90β was first assessed in vitro in lung epithelial cells, A549 cells and primary lung fibroblasts from control and IPF patients. While the secretion of HSP90α was upregulated without any stimulation in IPF fibroblasts compared with controls, A549 cells did not secrete any HSP90α even after stimulation with TGF-β1 (figure 2a). These results were confirmed by Western blot analysis on supernatant of primary fibroblasts and A549 cells (supplementary figure S3). No HSP90β secretion was observed in A549 cells or primary fibroblasts (figure 2a and supplementary figure S3). Regular tissue culture plates are very stiff (range of 1 GPa) compared with normal lung tissue (range of 1 kPa). Therefore, we cultured our primary fibroblasts on hydrogels of various stiffness in order to measure HSP90α secretion. Soft hydrogels had a stiffness of 1 kPa (corresponding to a normal lung) and stiff hydrogels had a stiffness of 50 kPa (corresponding to a fibrotic lung) [7]. On stiff hydrogels and regular tissue culture plates there was an upregulation of HSP90α secretion in IPF fibroblasts compared with controls. However, secretion of HSP90α by IPF fibroblasts was inhibited on soft substrate (figure 2b). Primary fibroblasts did not secrete HSP90β on soft or stiff hydrogels (figure 2b). Another important stimulus linked with disease progression in IPF is mechanical stretch. As the disease progresses, the increase in ECM deposition induces an increase in lung tissue stiffness, which in turn aggravates mechanical stretch during breathing and favours IPF progression [22]. Our team developed a model that mimics mechanical stretch that occurs during breathing in IPF patients (supplementary figure S1). This model allows the application of mechanical stretch to a fibrotic lung slice and the measurement of ex vivo tissue stiffness and quantification of secreted proteins after mechanical stretch stimulus [22]. Using this model, we demonstrated that mechanical stretch induced the release of HSP90α, but not HSP90β, from fibrotic lung slices of AdTGF-β1-treated rats, whereas nonfibrotic lung slices did not release HSP90α or HSP90β (figure 2c). Moreover, the release of HSP90α was positively correlated with the stiffness (Young's modulus) of fibrotic lung slices (supplementary figure S3). Interestingly, pre-treatment of the fibrotic lung slices with an inhibitor of cellular secretion completely abrogated the release of HSP90α induced by mechanical stretch (figure 2d).

Mechanical stretch and tissue stiffness induce heat shock protein (HSP) isoform HSP90α secretion. a) Level of HSP90α and HSP90β in supernatant of untreated control/primary idiopathic pulmonary fibrosis (IPF) lung fibroblasts and A549 cells treated or not with recombinant transforming growth factor (rTGF)-β1. HSP90α and HSP90β were measured by ELISA after cells were placed in serum-free medium for 24 h. Data presented as mean±sem; n=3. b) Western blot analysis of HSP90α and HSP90β expression in supernatant (with corresponding densitometry analysis for HSP90α). Cells were cultured either on hydrogels of stiffness 1 kPa (soft) or 50 kPa (stiff) or on regular tissue culture plates (1 GPa) and placed in serum-free medium for 24 h before supernatant collection. Ponceau staining served as loading control. Data presented as mean±sem; n=4. c) Western blot analysis of HSP90α and HSP90β expression release by nonfibrotic and fibrotic lung slices (from adenovector-mediated transforming growth factor-β1 (AdTGF-β1)-treated rats) before and after stretch. Western blots are representative of three independent experiments. Western blots of fibrotic and nonfibrotic lung slices were performed on separated gels but run simultaneously. Ponceau staining served as loading control. d) Western blot analysis of HSP90α and HSP90β expression release by fibrotic lung slices (from AdTGF-β1-treated rats) before and after stretch with or without protein transport inhibitor (PTI) treatment (with corresponding densitometry analysis for HSP90α). S: stimulated; US: unstimulated. Ponceau staining served as loading control. Data presented as mean±sem; n=6. *: p<0.05; **: p<0.01; ***: p<0.001.
Taken together, these results demonstrate that HSP90α, but not HSP90β, is secreted from IPF lung fibroblasts after stimulation by tissue stiffness and mechanical stretch.
Extracellular HSP90α induces myofibroblast differentiation
The treatment of lung epithelial cells and control fibroblasts with rHSP90α induced an upregulation of the major pro-fibrotic markers. Indeed, rHSP90α promoted the expression of α-SMA, the marker of myofibroblasts, and also induced the TGF-β1 pathway via Smad2 phosphorylation as well as the ERK pathway. In turn, rHSP90α enhanced the production of collagen 1A (figure 3a and b). Interestingly, the upregulation of α-SMA and downregulation of the epithelial marker E-cadherin by rHSP90α in A549 cells suggests induction of EMT (figure 3a). The treatment of IPF fibroblasts with rHSP90α did not induce changes in the expression of pro-fibrotic markers, which were already highly expressed (supplementary figure S4). In contrast, the inhibition of HSP90α with a blocking antibody reduced α-SMA and collagen 1A expression as well as ERK phosphorylation in IPF fibroblasts (figure 3c). Interestingly, conditioned medium from IPF fibroblasts, but not from control fibroblasts, favoured epithelial cell migration. This migration was hampered by the inhibition of HSP90α with a blocking antibody (figure 3d).

Extracellular heat shock protein (HSP) isoform HSP90α induces myofibroblast differentiation. a) Western blot analysis (and corresponding densitometry) of E-cadherin, collagen 1A, phosphorylated extracellular signal-regulated kinase (p-ERK), ERK, α-smooth muscle actin (α-SMA), phosphorylated Smad2 (p-Smad2) and Smad2 on A549 cells treated with recombinant (r) HSP90α or vehicle (control) for 48 h at 10 µM. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) staining served as loading control. Data presented as mean±sem; n=4. b) Western blot analysis (and corresponding densitometry) of collagen 1A, p-ERK, ERK, α-SMA, p-Smad2 and Smad2 on human primary control fibroblasts treated with rHSP90α or vehicle (control) for 48 h at 10 µM. GAPDH staining served as loading control. Data presented as mean±sem; n=4. c) Western blot analysis (and corresponding densitometry) of collagen 1A, p-ERK, ERK and α-SMA on human primary idiopathic pulmonary fibrosis (IPF) fibroblasts treated with a blocking HSP90α antibody (AbHSP90α) or vehicle (control) for 48 h at 40 µM. GAPDH staining served as loading control. Data presented as mean±sem; n=4. d) Representative images of the wound closure assay performed on A549 cells cultured with conditioned medium (CM) from control or IPF primary fibroblasts and treated with AbHSP90α or vehicle (control) at 40 µM. Images are representative of three independent experiments. The graph represents the percentage of wound closure measured at 36 h after the wound was treated. Corresponding proliferating cell nuclear antigen (PCNA) expression of A549 cells showed no increase in proliferation during the wound closure assay. Data presented as mean±sem; n=3. *: p<0.05; **: p<0.01; ***: p<0.001.
Taken together, these results demonstrate that HSP90α promotes EMT of lung epithelial cells and differentiation of fibroblasts into myofibroblasts, whereas its inhibition limits the aggressive phenotype of IPF fibroblasts.
HSP90α signals through the receptor LRP1
It has been shown that extracellular HSP90 activity is driven by the receptor LRP1 [15]. Surprisingly, we were not able to co-immunoprecipitate HSP90α and LRP1 from plasma membrane protein extracts of control or IPF fibroblasts (figure 4a). However, HSP90β and LRP1 co-immunoprecipitated strongly (figure 4a). We hypothesised that the extracellular binding between HSP90α and LRP1 might be too weak to show a positive co-immunoprecipitation. Therefore, we pre-treated our cells with a protein cross-linker before performing the co-immunoprecipitation. In control fibroblasts, HSP90α and LRP1 did not co-immunoprecipitate without or with prior cross-linking (figure 4b), whereas HSP90α and LRP1 co-immunoprecipitated in IPF fibroblasts after cross-linking (figure 4b). Therefore, we hypothesised that the binding between HSP90α and LRP1 was only extracellular (only visible after cross-linking), whereas there was also an unexpected binding of HSP90β and LRP1 from inside the cell (strong co-immunoprecipitation without cross-linking). As the intracellular role of HSP90β is mainly protein chaperoning (protection from degradation), we inhibited intracellular HSP90β in IPF fibroblasts using specific siRNA. The inhibition of HSP90β induced a strong downregulation of LRP1 expression. The expression of LRP1 was restored when cells where treated with MG132, an inhibitor of the proteasome degradation pathway (figure 4c). However, intracellular inhibition of HSP90α did not induce LRP1 downregulation (figure 4c).

Heat shock protein (HSP) isoform HSP90α signals through low-density lipoprotein receptor-related protein 1 (LRP1). a) Immunoprecipitation (IP) of HSP90α and HSP90β performed on plasma membrane extracted proteins followed by immunodetection of LRP1 and HSP90α. Control: control human primary fibroblasts; IPF: idiopathic pulmonary fibrosis human primary fibroblasts; IgG: nonrelevant antibody; input: nonimmunoprecipitated extracts. Na,K-ATPase served as loading control of the plasma membrane fraction. b) Immunoprecipitation of LRP1 performed on plasma membrane extracted proteins followed by immunodetection of HSP90α, HSP90β and LRP1. Immunoprecipitation was performed with or without prior cross-linking of extracellular/membrane proteins: control human primary fibroblasts and human primary IPF fibroblasts. NC: non-cross-linked; C: cross-linked. c) Western blot analysis of LRP1, HSP90α and HSP90β expression on IPF human primary fibroblasts treated with small interfering (si) RNA for HSP90α or HSP90β (Scramble siRNA used as control) and treated with or without MG132 for 6 h at 50 µM. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) staining served as loading control. The Western blot is representative of three independent experiments. d) Western blot analysis of phosphorylated extracellular signal-regulated kinase (p-ERK), ERK, α-smooth muscle actin (α-SMA), phosphorylated Smad2 (p-Smad2), Smad2, LRP1 and collagen 1A expression on human primary IPF fibroblasts, A549 cells and control fibroblasts treated with siRNA for LRP1 (Scramble siRNA used as control) and treated with recombinant (r) HSP90α for 48 h at 10 µM. GAPDH staining served as loading control. Data presented as mean±sem; n=3. *: p<0.05; **: p<0.01; ***: p<0.001.
In order to confirm the signalling pathway involving HSP90α and LRP1, lung epithelial cells, control and IPF fibroblasts were treated with rHSP90α, while LRP1 was inhibited via a siRNA. Interestingly, LRP1 inhibition completely abrogated the effect of rHSP90α in all cell types by preventing α-SMA and collagen upregulation and Smad2 and ERK phosphorylation (figure 4d).
Taken together, these results demonstrate that HSP90α signals through LRP1 and that intracellularly enhances LRP1 stability.
HS-30, an inhibitor of extracellular HSP90, prevents HSP90α/LRP1 signalling
HS-30 is a selective inhibitor of HSP90 that has been modified to be cell impermeable [20]. Treatment of control and IPF fibroblasts with HS-30 inhibited the effects of rHSP90α on these cells by preventing α-SMA and collagen upregulation and Smad2 and ERK phosphorylation (figure 5a and b). Interestingly, HS-30 inhibited the co-immunoprecipitation of HSP90α and LRP1 in cross-linked IPF fibroblasts (figure 5c). This result was confirmed by immunofluorescence. The colocalisation of HSP90α and LRP1 was enhanced in IPF fibroblasts compared with control fibroblasts. HS-30 significantly inhibited HSP90α and LRP1 colocalisation (figure 5d). To further confirm the link between HSP90α and LRP1 in humans we performed immunofluorescence on human lung tissue. HSP90α and LRP1 colocalisation was significantly increased in patients with moderate and severe IPF compared with controls (figure 6).

HS-30, an inhibitor of extracellular heat shock protein HSP90, prevents HSP90α/low-density lipoprotein receptor-related protein 1 (LRP1) signalling. a) Western blot analysis of collagen 1A, α-smooth muscle actin (α-SMA), phosphorylated extracellular signal-regulated kinase (p-ERK), ERK, HSP90α and LRP1 expression on human primary control fibroblasts treated with recombinant (r) HSP90α for 48 h at 10 µM with or without HS-30 at 1 mM. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) staining served as loading control. The Western blot is representative of three independent experiments. b) Western blot analysis of α-SMA, p-ERK, ERK, phosphorylated Smad2 (p-Smad2), Smad2, HSP90α and LRP1 expression on human primary idiopathic pulmonary fibrosis (IPF) fibroblasts treated with rHSP90α for 48 h at 10 µM with or without HS-30 at 1 mM. GAPDH staining served as loading control. The Western blot is representative of three independent experiments. c) Immunoprecipitation (IP) of LRP1 performed on plasma membrane extracted proteins followed by immunodetection of HSP90α and LRP1. C: cross-linked; input: nonimmunoprecipitated extracts; IgG: nonrelevant antibody. Immunoprecipitation was performed with prior cross-linking of extracellular/membrane proteins. Immunoprecipitation performed on IPF fibroblasts treated with or without HS-30 at 1 mM. d) Immunofluorescence of LRP1 (red) and HSP90α (green) on control fibroblasts, IPF fibroblasts and IPF fibroblasts treated with HS-30 at 1 mM. Nuclear staining: 4′,6-diamidino-2-phenylindole (blue). Images are representative of three independent experiments. Scale bar: 50 μm. Pearson's coefficient for each condition presented as mean±sem; n=3. **: p<0.01; ***: p<0.001.

Heat shock protein (HSP) isoform HSP90α/low-density lipoprotein receptor-related protein 1 (LRP1) colocalise in idiopathic pulmonary fibrosis (IPF) patients. Immunofluorescence of LRP1 (red) and HSP90α (green) on human lung tissue from controls, moderate IPF patients and severe IPF patients. IgG as inserts. Nuclear staining: 4′,6-diamidino-2-phenylindole (blue). Images presented are representative of three independent experiments. Scale bar: 50 μm. Pearson's coefficient for each condition is presented as mean±sem; n=3. ***: p<0.001.
HSP90α signalling in IPF fibroblasts is independent of the TGF-β1 pathway
Extracellular HSP90α has recently been shown to bind TGF-βRI, promoting collagen production in TGF-β-activated cardiac fibroblasts [25]. In our model, extracellular HSP90α did not co-immunoprecipitate with TGF-βRI in IPF fibroblasts even after cross-linking (supplementary figure S5). Moreover, SD-208, a TGF-βRI inhibitor, did not inhibit the increase in α-SMA and TGF-β1 or the phosphorylation of ERK induced by rHSP90α in lung fibroblasts (supplementary figure S6). Similarly, rHSP90α induces an increase in TGF-β1 expression and activation that was not inhibited by SD-208, while it was inhibited by HS-30 (supplementary figure S6).
Taken together, these results demonstrate that HSP90α signalling is independent of the TGF-β1 pathway.
HS-30 has antifibrotic properties on fibrotic lung tissue ex vivo
Fibrotic lung slices from day 21 AdTGF-β1-treated animals were treated with HS-30 or vehicle for 72 h ex vivo. HS-30 induced a decrease in mRNA expression of collagen and α-SMA in lung slices from AdTGF-β1-treated animals compared with controls (figure 7a). In addition, HS-30 also decreased the protein expression of α-SMA and TGF-β1, demonstrating a role of the inhibition of extracellular HSP90 on myofibroblast differentiation and persistence in fibrotic lung tissue (figure 7b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HS-30 has antifibrotic properties on fibrotic lung tissue ex vivo. a) Quantitative reverse transcription PCR analysis of α-smooth muscle actin (α-SMA) and collagen 1A expression on lung slices from rats receiving adenovector-mediated transforming growth factor-β1 (AdTGF-β1). Lung slices were collected at day 21 after AdTGF-β1 administration and then cultured ex vivo with HS-30 or vehicle for 72 h. Data presented as mean±sem; n=4. b) Western blot analysis (and corresponding densitometry) of α-SMA and TGF-β1 expression on lung slices from rats receiving AdTGF-β1. Lung slices were collected at day 21 after AdTGF-β1 administration and then cultured ex vivo with HS-30 or vehicle for 72 h. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) staining served as loading control. Data presented as mean±sem; n=4. *: p<0.05; **: p<0.01.
Discussion
IPF is characterised by the emergence of apoptosis-resistant myofibroblasts that excessively produce abnormal ECM responsible for lung tissue stiffening and disease progression. Thus, IPF is viewed as an uncontrolled wound healing response in which numerous factors hamper the clearance of myofibroblasts necessary to form proper scar tissue. In the case of IPF, the persistence of myofibroblasts allows scar tissue accumulation, progressively destroying normal lung architecture and compromising lung function [26]. The central role of TGF-β1 in myofibroblast differentiation has been documented repeatedly [6, 8]. In the current study we identified another circulating protein, HSP90α, which is able to induce myofibroblast differentiation from lung epithelial cells and lung fibroblasts. Interestingly, the circulating HSP90α level correlated with collagen deposition in an animal model of lung fibrosis and also correlated with disease severity in patients with IPF. Further studies with larger numbers of IPF patients and patients with other non-IPF interstitial lung diseases are required to assess the potential of circulating HSP90α as a biomarker for IPF severity and IPF progression in patients.
Our results highlight that the secretion of pro-fibrotic HSP90α is driven by tissue stiffness and mechanical stretch. As a result of the accumulation of scar tissue, IPF lungs are stiffer than normal lungs [7]. Therefore, IPF lungs are subjected to higher mechanical stretch during breathing [23]. The increase in stiffness and mechanical stretch forces in IPF lung have already been shown to be involved in fibrosis progression, mainly by TGF-β1 activation that in turn activates myofibroblasts to produce more abnormal ECM [22]. In our study we demonstrate that, along with TGF-β1 activation, mechanical stretch applied to fibrotic lung slices induces the release of HSP90α into the extracellular space, which is also able to activate myofibroblast differentiation. We further demonstrate that HSP90α signals through the receptor LRP1 to promote myofibroblast differentiation. Surprisingly, while HSP90β is not secreted, it binds to LRP1 intracellularly, thus stabilising the receptor and further activating HSP90α/LRP1 signalling. These findings are in accordance with results obtained in a wound healing model in which extracellular HSP90 has been shown to signal through LRP1 in promoting skin cell migration in vitro and in vivo [27]. Moreover, Jayaprakash et al. [28] demonstrated that HSP90α and HSP90β work together in promoting cell motility in wounded skin and thereby accelerate wound closure. In their model, HSP90β stabilised LRP1, while the secreted HSP90α signalled through LRP1 [28]. Very recently, Sontake et al. [29] demonstrated distinct roles for HSP90α and HSP90β in IPF. In their study, the specific intracellular inhibition of HSP90β attenuated pro-fibrotic gene expression, such as the genes for collagen 1α, collagen 5α and α-SMA. Interestingly, the authors demonstrated that intracellular inhibition of HSP90α did not have an effect on pro-fibrotic signalling pathways. These results are in line with our study in which we clearly demonstrate that the pro-fibrotic effects of HSP90β are mediated inside the cell, whereas they are extracellular for HSP90α. In IPF, the increased stiffness of the lung tissue might overactivate this pathway by stimulating HSP90α secretion, leading to uncontrolled wound healing and progressive fibrosis. These results suggest that both oversecretion of HSP90α and increased lung stiffness are sources for constantly active HSP90α as a pro-fibrotic signal. Therefore, it would be an effective therapeutic strategy to concomitantly inhibit extracellular HSP90α and reduce lung stiffness. Such a strategy showed promising results in animal models of scleroderma-associated lung disease. The stiffness of IPF lung tissue is amplified by ECM cross-linking [30]. Lin et al. [31] demonstrated that a therapy against myofibroblast contractility using relaxin was effective only if it was combined with a therapy targeting matrix cross-linking in order to limit lung stiffness. One can imagine that inhibiting HSP90α extracellular signalling combined with the same kind of therapy aiming to reduce lung stiffness could represent a promising approach against already established fibrosis.
Interestingly, the HSP90α extracellular signalling that we describe in this study appears to be independent of the major pro-fibrotic pathway, TGF-β1. Intracellular HSP90 has been shown to stabilise TGF-βRII and promote myofibroblast differentiation in lung and kidney fibrosis [32–35]. Moreover, extracellular HSP90 has been recently shown to bind TGF-βRI and participate in TGF-β1-induced collagen production in myocardial fibroblasts [25]. However, in our model of primary human lung fibroblasts, the effect of extracellular HSP90α could not be hampered by SD-208, a specific inhibitor of TGF-βRI. As IPF fibroblasts are expressing high levels of LRP1 we believe that the HSP90α/LRP1 signalling drives, at least in part, myofibroblast persistence. The recent findings showing that HSP90 is upregulated in patients with IPF and that therapeutic administration of an HSP90 inhibitor, 17-N-allylamino-17-demethoxy-geldanamycin (17-AAG), inhibits bleomycin-induced lung fibrosis highlight the potential benefits of HSP90 inhibition in IPF [29]. However, 17-AAG is targeting HSP90α and HSP90β intracellular activity. With regard to the results presented here, we believe that specifically targeting extracellular HSP90α signalling in IPF could help to overcome possible issues resulting from a complete inhibition of the cytoprotective function of intra- and extracellular HSP90. There is no specific in vivo inhibitor of extracellular HSP90α available, which limits our study somewhat in fully characterising the in vivo effects of extracellular HSP90 inhibition during lung fibrosis. Nevertheless, our results demonstrate on fibrotic lung tissue from rats that the inhibition of extracellular HSP90 shows promising antifibrotic effects by reducing collagen expression and myofibroblast differentiation and persistence ex vivo. Therefore, this new experimental study together with an increasing body of evidence from experimental and clinical research puts HSPs, and especially HSP90α and HSP90β, more and more into the focus of drug development for IPF.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00386-2017_Supplement
Figure S1. Mechanical stretch machine. a) Top-down schematic representation of the ex vivo model to apply mechanical stretch to lung strips. b) Photograph of the ex vivo model to apply mechanical stretch. ERJ-00386-2017_Figure_S1
Figure S2. Circulating HSP90α is upregulated in IPF patients and in AdTGF-β1 induced lung fibrosis. a) Serum level of HSP90β measured by ELISA on IPF patients and respective aged-matched healthy volunteers (Left panel). Results are presented as mean±SEM, n=9 and n=31 for controls and IPF patients respectively. IPF patients have been clustered in 2 groups (right panel): moderate IPF (FVC % predicted >60%) and severe IPF (FVC % predicted <60%). Results are presented as mean±SEM. ***p<0.001, n=9, n=25 and n=6 for controls, moderate IPF and severe IPF patients respectively. b) Western blot analysis of HSP90 expression in BALF of rats treated with AdTGF-β1 (AdDL=control). WB presented is representative of 3 independent experiments. c) Correlation curves between the serum and BALF level of HSP90α in rats treated with AdTGF-β1 and Ashcroft scores of corresponding lungs. ERJ-00386-2017_Figure_S2
Figure S3. Mechanical stretch and tissue stiffness induce HSP90α secretion. A. Western blot analysis of HSP90α and HSP90β expression in cells’ supernatant of untreated control/IPF primary lung fibroblasts and A549 treated or not with rTGF-β1. WB presented are representative of 3 independent experiments. Ponceau staining served as loading control. b) Correlation curves between the levels of HSP90α released by fibrotic lung strips after stretch and Youngâs modulus (stiffness) of corresponding lung strips. ERJ-00386-2017_Figure_S3
Figure S4. HSP90α does not induce pro-fibrotic markers in IPF fibroblasts Western blot analysis (and corresponding densitometry) of collagen 1A, p-ERK, ERK, p-Smad2, Smad2 and α-SMA on human primary IPF fibroblasts treated with rHSP90α or vehicle (ctrl) for 48 h at 10 μM. GAPDH staining served as loading control. Results are presented as mean±SEM, n=4. ERJ-00386-2017_Figure_S4
Figure S5. rHSP90α does not interact with TGFβRI. a) Immunoprecipitation (IP) of HSP90α performed on plasma membrane extracted proteins followed by immuno-detection of TGFβRI and HSP90α. IP was performed with prior crosslinking of extracellular/membrane proteins. IP performed on IPF fibroblasts treated with or without rHSP90α for 48 h at 10 μM. IgG: non-relevant antibody; input: non-immunoprecipitated extracts. ERJ-00386-2017_Figure_S5
Figure S6. HSP90α signaling in IPF fibroblasts is independent of TGF-β1 pathway. a) Western blot analysis of TGF-β1, α-SMA, p-ERK and ERK expression on human primary control fibroblasts treated with rHSP90α for 48 h at 10 μM and with or without SD-208, an ALK5 inhibitor at 30 μM. GAPDH staining served as loading control. WB presented is representative of 3 independent experiments. b) Total and active TGF-β1 levels measured by ELISA on control fibroblasts treated with rHSP90α for 48 h at 10 μM and with or without SD-208, an ALK5 inhibitor at 30 μM. Results are presented as mean±SEM, **p<0.01, ***p<0.001, n = 5. c) Total and active TGF-β1 levels measured by ELISA on control fibroblasts treated with rHSP90α for 48 h at 10 μM and with or without HS-30 at 1 mM. Results are presented as mean±SEM, **p<0.01, ***p<0.001, n = 5. ERJ-00386-2017_Figure_S6
Disclosures
Supplementary Material
T. Haystead ERJ-00386-2017_Haystead
P. Hughes ERJ-00386-2017_Hughes
M. Kolb ERJ-00386-2017_Kolb
Acknowledgements
The authors would like to thank Mara Ludwig (McGill University, Montreal, ON, Canada) for her help with tissue bath apparatus designs.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: C. Shimbori is funded by the Pulmonary Fibrosis Foundation (I.M. Rosenzweig Junior Investigator Award). P-S. Bellaye is funded by le Fonds de Dotation “Recherche en Santé Respiratoire” et de la Fondation du Souffle, the Canadian Institutes of Health Research and the Canadian Pulmonary Fibrosis Foundation. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received February 23, 2017.
- Accepted November 7, 2017.
- Copyright ©ERS 2018
References










