





Abstract
Background Idiopathic pulmonary arterial hypertension (IPAH) is a rare disease with high heritability. Although several predisposing genes have been linked to IPAH, the genetic aetiology remains unknown for a large number of IPAH cases.
Methods We conducted an exome-wide gene-based burden analysis on two independent case–control studies, including a total of 331 IPAH cases and 10 508 controls. Functional assessments were conducted to analyse the effects of genetic mutations on protein biosynthesis and function.
Results The gene encoding human bone morphogenetic protein 9 (BMP9) was identified as a novel genetic locus displaying exome-wide association with IPAH in the discovery cohort (OR 18.8; p=1.9×10−11). This association was authenticated in the independent replication cohort (p=1.0×10−5). Collectively, the rare coding mutations in BMP9 occurred in 6.7% of cases, ranking this gene second to BMPR2, comprising a combined significance of 2.7×10−19 (OR 21.2). Intriguingly, the patients with BMP9 mutations had lower plasma levels of BMP9 than those without. Functional studies showed that the BMP9 mutations led to reduced BMP9 secretion and impaired anti-apoptosis ability in pulmonary arterial endothelial cells.
Conclusion We identify BMP9 as an IPAH culprit gene.
Abstract
BMP9 is a new culprit gene for IPAH ranking second to BMPR2. The rare deleterious mutations in BMP9, which lead to the reduction in BMP9 secretion and impairment in BMP9 function, account for 6.7% of IPAH cases. http://ow.ly/h0tS30mXr8j
Introduction
Idiopathic pulmonary arterial hypertension (IPAH), which accounts for 35–67% of pulmonary arterial hypertension (PAH) cases, is a rare but fatal disease with high heritability [1–4]. The genetic basis of IPAH has been studied extensively. Rare coding mutations in bone morphogenetic protein receptor type 2 (BMPR2) are found in ∼70% of familial kindreds with PAH and 15–20% of sporadic IPAH cases [5–7]. Less frequently, rare mutations of large effect are present in the genes encoding caveolin-1 (CAV1), potassium channel two-pore domain subfamily K member 3 (KCNK3), SMAD family member 1 (SMAD1), SMAD family member 9 (SMAD9), T-box 4 (TBX4) and bone morphogenetic protein receptor type 1B (BMPR1B) [8–13]. Additionally, activin A receptor-like type 1 (ACVRL1) and endoglin (ENG) were identified as genetic loci implicated in the development of both PAH and hereditary haemorrhagic telangiectasia (HHT) [14, 15]. The biallelic recessive mutations in eukaryotic translation initiation factor 2α kinase 4 (EIF2AK4) were shown recently to be a major cause of pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis, which are clinically and pathologically distinct forms of PAH [16]. Collectively, the mutations of the 10 genes could only explain a fraction (∼20%) of IPAH cases, suggesting additional IPAH genes must exist [17].
The canonical approach to identify IPAH susceptibility genes involves the use of family-based genetics. To date, data generated from multigenerational PAH families have been commonly used to reveal mutations in one of these known PAH genes. The scarcity of IPAH makes it more difficult to identify new IPAH-related genes via targeting a larger number of cases for nonbiased whole-genome screening. Moreover, PAH stems from an interplay among many detrimental factors, including, but not limited to, drugs, environmental cues, age, sex and ethnic background. Thus, the genotypic data would not be the sole confounding factor for the clinical heterogeneity. With the aforementioned limitations, a large-scale analysis of genetic specimens with well-defined IPAH cases in conjunction with state-of-the-art methods is urgently needed to identify other IPAH susceptibility loci.
The analysis of data from next-generation sequencing (NGS) enables the identification of novel disease genes. Here, we aimed to identify IPAH causative genes by performing two-stage NGS in sporadic IPAH cases enrolled in two centres in China for >10 years. While the extent of the previously identified PAH genes (e.g. BMPR2) was verified, gene-based burden analysis was performed to screen IPAH susceptibility genes.
Methods
A detailed description of all methods can be found in the supplementary material.
Study design
IPAH patients were recruited from two national pulmonary hypertension referral centres in China: Shanghai Pulmonary Hospital of Tongji University in Shanghai (with patients mainly from south China) and FuWai Hospital of the Chinese Academy of Medical Sciences in Beijing (with patients mainly from north China). The study comprised two stages: “discovery” and “replication” stages. All of the IPAH cases were genotyped by NGS and screened for rare variants in 10 previously reported PAH genes (BMPR2, ACVRL1, ENG, SMAD1, SMAD9, TBX4, BMPR1B, CAV1, KCNK3 and EIF2AK4). Gene-based burden analysis was then performed by comparing the exome data of cases against controls to identify novel IPAH susceptibility genes. These putative genes identified in the discovery cohort were subsequently evaluated in the replication cohort. The correlation between genotype and clinical phenotype was further analysed with the use of combined datasets. The research protocols were approved by the institutional review boards in Shanghai Pulmonary Hospital and FuWai Hospital. The study design is shown in figure 1.

Flowchart of study design. PAH: pulmonary arterial hypertension; IPAH: idiopathic pulmonary arterial hypertension; APVT: acute pulmonary vasoreactivity testing; HHT: hereditary haemorrhagic telangiectasia; NGS: next-generation sequencing; WES: whole-exome sequencing; gnomAD: Genome Aggregation Database.
Statistical analysis
Results are reported as percentage, median (interquartile range (IQR)) or mean with standard deviation, as indicated. The normality of data distribution was assessed using Kolmogorov–Smirnov tests. A Chi-squared test or Fisher's exact test was applied to compare qualitative variables and genotype/allele frequencies. For quantitative variables, statistical significance was determined using an unpaired t-test or one-way ANOVA. The Mann–Whitney U-test or Kruskal–Wallis test was performed to analyse nonnormally distributed variables. All statistical tests were two-tailed. Results were considered statistically significant at a level of p<0.05. All analyses were performed with PASW Statistics version 18.0 (SPSS, Chicago, IL, USA).
Results
Study population
A total of 331 patients with IPAH were recruited from two national referral centres (figure 1). For the discovery cohort, 122 cases (15 paediatric patients) were recruited from Shanghai Pulmonary Hospital and 129 cases (33 paediatric patients) were recruited from FuWai Hospital. For the replication cohort, 80 incident cases (nine paediatric patients) were enrolled from FuWai Hospital. Demographic and clinical characteristics of the two IPAH cohorts are described in table 1.
Demographic characteristics of the patients in each cohort and the prevalence of bone morphogenetic protein 9 (BMP9) mutations
Identification of BMP9 as an IPAH susceptibility gene
In the discovery stage, we genotyped 36 IPAH cases using whole-genome sequencing (WGS) and the other 215 cases using whole-exome sequencing (WES). WGS and WES achieved an average coverage of 37- and 128-fold on target, respectively. More than 92% of targeted bases were sequenced to a read depth of more than 20 times (supplementary table S1).
We first analysed the data for rare variants and mutations in the 10 previously reported PAH-predisposing genes. The rare heterozygous mutations of BMPR2 were found in 49 cases (19.5%). For the other PAH risk genes, we identified deleterious rare variants in ACVRL1 (15 cases (6%)), TBX4 (10 cases (4%)), SMAD1 (two cases (0.8%)), BMPR1B (one case (0.4%)), KCNK3 (one case (0.4%)) and SMAD9 (one case (0.4%)). Four cases (1.6%) harboured biallelic EIF2AK4 mutations: two cases carried homozygous mutations and two cases carried heterozygous mutations (one missense and one frameshift in one case, and two missense mutations in the other case). Eight cases (3.2%) had double heterozygous mutations or rare variants in two different PAH risk genes. In total, rare variants and mutations were identified in 91 patients, corresponding to 36.3% of all cases (supplementary table S2). The details for all rare variants and mutations of the 10 PAH genes are provided in supplementary dataset S1.
To this end, the identified rare variants and mutations in genes known to be IPAH related confirmed the genetic background of the discovery cohort. We then performed a gene-based burden analysis utilising the exome data of 251 cases and 1884 controls (supplementary figure S1). Further analysis of the homogenous population structure demonstrated that all cases and controls were clustered with subjects of Chinese ancestry in the 1000 Genomes dataset (www.1000genomes.org) (supplementary figure S2). A total of 13 318 genes had more than one case in 251 IPAH patients with a potential deleterious genetic variant for the dominant model tested. After normalisation for multiple tests (p<3.8×10−6 after accounting for the 13 318 genes examined), only three genes (BMPR2, BMP9 (encoding bone morphogenetic protein 9) and ACVRL1) had an exome-wide significant enrichment of mutations in IPAH cases when compared with controls (table 2 and supplementary figure S3). Noticeably, besides the well-documented PAH causative BMPR2 and ACVRL1, BMP9 would be a new gene displaying exome-wide association with IPAH. Mutations in BMP9 were found in 6.8% (17 out of 251) of cases and 0.4% (seven out of 1884) of controls (OR 18.8, 95% CI 7.4–53.9; raw p=1.9×10−11, Bonferroni corrected p=2.6×10−7). Under the dominant coding model, BMP9 ranked second next to BMPR2 (table 2 and supplementary figure S3). Specifically, BMP9 mutations were found in 5.7% (seven out of 122) and 7.8% (10 out of 129) of cases from Shanghai Pulmonary Hospital and FuWai Hospital, respectively (table 1). The distribution of BMP9 mutations did not significantly differ between the two centres (p=0.53, Chi-squared test).
Association of three genes with idiopathic pulmonary arterial hypertension (IPAH) in two independent case–control studies
To further validate the risk effects of BMP9, we performed WES in an independent replication cohort of 80 IPAH cases. Rare variants and mutations in PAH-related genes were identified in 37 (46.3%) cases (summary in supplementary table S2; all rare variants and mutations are listed in supplementary dataset S1). Using the same pipeline of genetic burden analysis, we compared the 80 exomes of cases with the reference control exome sequencing data of 8624 East Asians from the Genome Aggregation Database (gnomAD-EAS; http://gnomad.broadinstitute.org). BMPR2, BMP9 and ACVRL1 were again identified as the top three disease-associated genes in the replication cohort (table 2). We identified five additional (6.3%) BMP9 heterozygous mutations (table 1 and supplementary table S3). The prevalence of BMP9 mutations was significantly higher than the reference controls of gnomAD-EAS (raw p=1.0×10−5). In the combined discovery and replication datasets, mutations in the BMP9 were found in 6.7% (22 out of 331) and 0.3% (34 out of 10 508) of cases and controls, respectively, comprising a combined significance of 2.7×10−19 (OR 21.2, 95% CI 11.7–37.6). The prevalence of BMP9 mutations was comparable between the discovery and replication cohorts (6.8% versus 6.3%). Therefore, this two-stage analysis strongly suggests that the BMP9 locus is a novel contributor to the pathogenesis of IPAH.
BMP9 mutation characteristics
Among the 331 IPAH patients, we identified 22 cases carrying 21 distinct rare heterozygous mutations in BMP9 (supplementary table S3). These mutations were spread throughout BMP9 (figure 2a and b). All the BMP9 mutations were confirmed by Sanger sequencing (supplementary figure S4). None of the BMP9 mutation carriers had any deleterious mutations in the 10 previously reported PAH genes. 20 of these BMP9 mutations were absent from more than 6512 exomes or genomes, including 301 Chinese samples from the 1000 Genome project, 1884 Novo-Zhonghua controls and 4327 East Asians in the Exome Aggregation Consortium database (ExAC-EAS; http://exac.broadinstitute.org). Only one mutation (p.G322R) was identified in these databases, with a prevalence of 0.0001–0.0002 (supplementary table S4). Of the 22 BMP9 mutation carriers, loss-of-function (LOF) mutations were found in six cases (27.3%), including three nonsense mutations, two frameshifts and one mutation that would affect the canonical translational start site (supplementary table S4). In the two reference populations, no such BMP9 mutations were found. The prevalence of BMP9 LOF mutations in IPAH cases (six cases (1.8%)) was significantly higher than in the two reference populations (p=1.1×10−5 for Novo-Zhonghua and p=2.5×10−9 for gnomAD-EAS controls). Regarding the 15 BMP9 missense mutations, in silico analyses suggested that these mutations would be deleterious (supplementary table S4) and located in the regions highly conserved among various species (figure 2c).
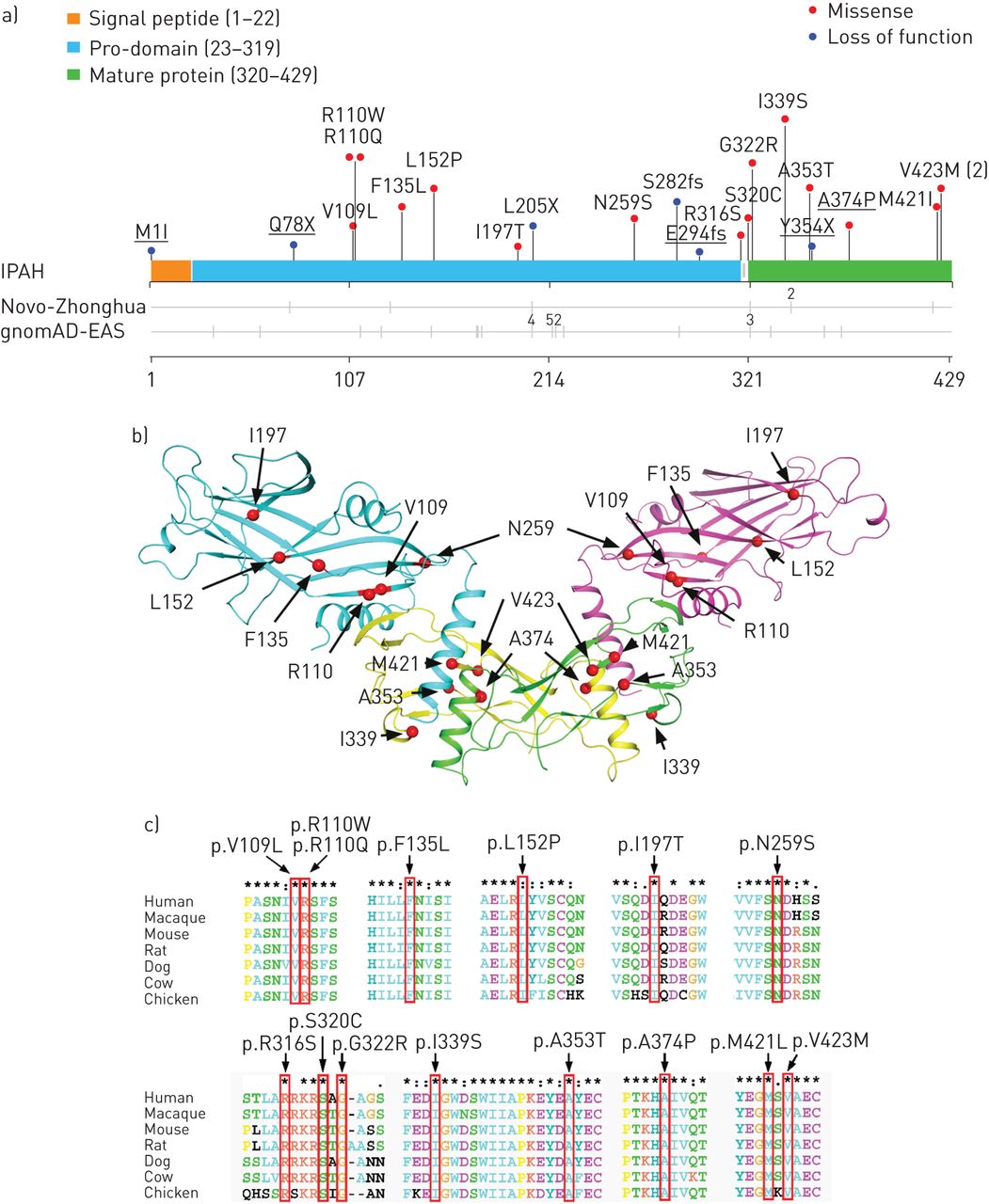
Bone morphogenetic protein 9 (BMP9) mutations in idiopathic pulmonary arterial hypertension (IPAH). gnomAD-EAS: Genome Aggregation Database East Asian. a) Spatial distribution of the mutations of BMP9 found in patients with IPAH, along with the distribution of variants in healthy controls. Mutations identified in the replication cohort are underlined. The numbers of subjects carrying the same mutation are indicated. b) Distribution of 11 BMP9 missense mutations that were found in IPAH patients displayed on the crystal structure of BMP9, with superimposition on growth factor dimers. The pro-domain ribbon is coloured blue and purple. The mature growth factor is coloured yellow and green. c) ClustalX (www.clustal.org) comparison of amino acid sequences shows high conservation of 15 missense BMP9 mutations across different species. The mutations are indicated in red boxes.
Clinical characteristics of BMP9 mutation carriers
Supplementary table S3 shows the clinical, functional and haemodynamic characteristics of the 22 IPAH patients carrying the various BMP9 mutations. Both males and females were affected, and a significant sex bias was evident with a female:male ratio of 4.5:1. Seven patients (31.8%) died during the follow-up.
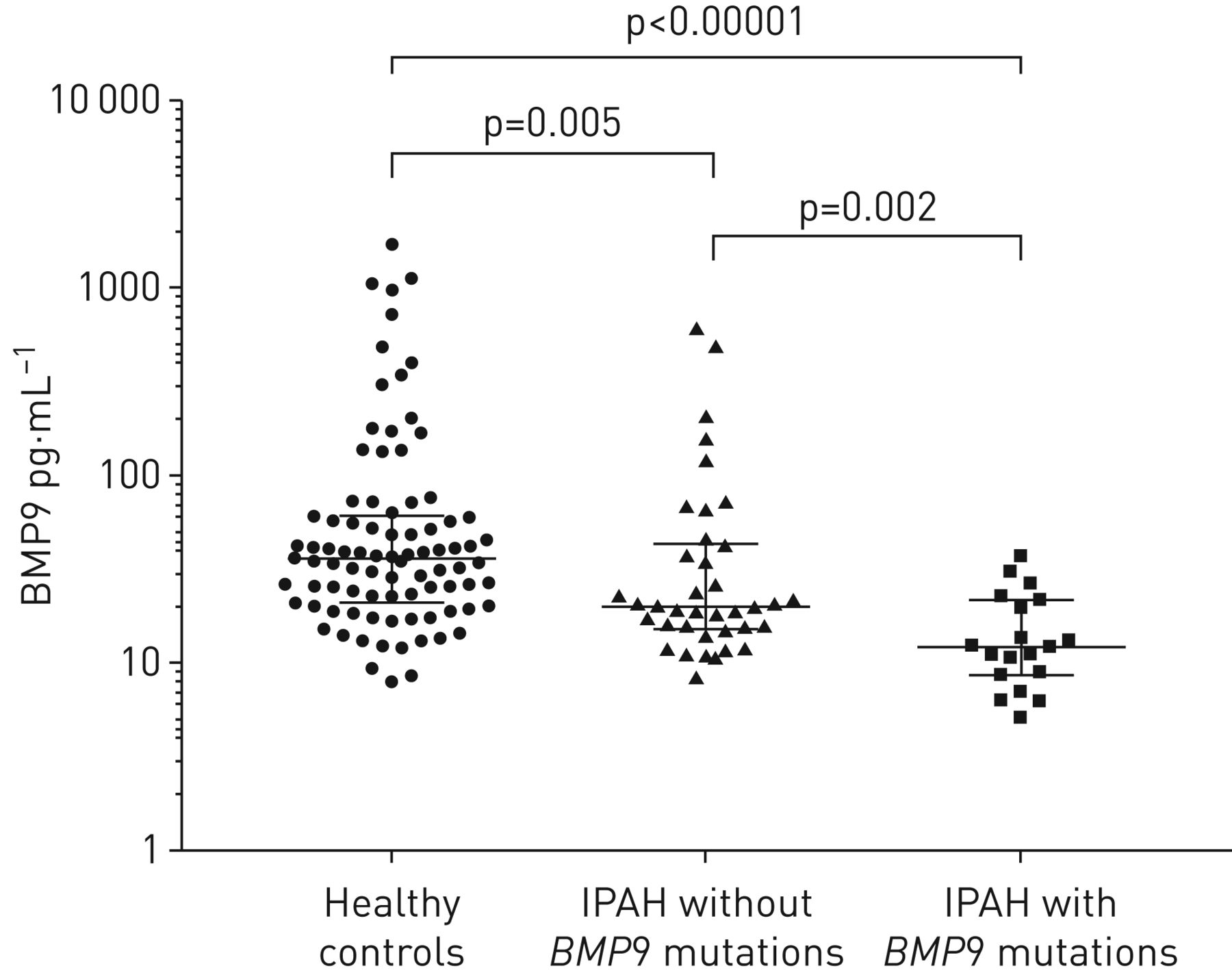
We expanded the analyses of BMP9 genotype and phenotypic outcomes in the combined datasets. Compared with the cases with or without BMP9 mutations, there were no significant differences in age and haemodynamics (supplementary table S5). As a member of the BMP subgroup of the transforming growth factor-β superfamily proteins, BMP9 is synthesised as a 429-amino-acid precursor protein (pre-pro-BMP9) and processed into pro-BMP9 (100 kDa) and mature BMP9 (25 kDa) (supplementary figure S5). The active form of BMP9 circulates in the blood and is crucial for the regulation of vasculature tone [18]. To investigate whether the BMP9 mutations affect the expression levels of BMP9, we analysed plasma BMP9 levels in IPAH patients carrying BMP9 mutations (n=19), age- and sex-matched healthy controls (n=87), and IPAH patients without BMP9 mutations (n=38) (supplementary table S6). The BMP9 levels were significantly different among the three groups (p<0.0001, Kruskal–Wallis test) (figure 3). Compared with healthy controls, the plasma BMP9 level was lower in IPAH patients lacking BMP9 mutations (median (IQR) 19.8 (15.2–43.4) versus 36.2 (20.8–60.7) pg·mL−1; p=0.005, Mann–Whitney test). Importantly, the BMP9 level was even lower in BMP9 mutation carriers when compared with patients without mutations (median (IQR) 12.1 (8.6–21.8) versus 19.8 (15.2–43.4) pg·mL−1; p=0.002, Mann–Whitney test) (figure 3). These results indicate that BMP9 mutations may suppress BMP9 synthesis and/or secretion, resulting in decreased circulating BMP9 levels that may be causative of IPAH.

Bone morphogenetic protein 9 (BMP9) levels in healthy controls (n=87), idiopathic pulmonary arterial hypertension (IPAH) patients without BMP9 mutations (n=38) and IPAH patients with BMP9 mutations (n=19). BMP9 was decreased in the IPAH patients without BMP9 mutations and lowest in BMP9 mutation carriers. The data are presented as individual data points with median and interquartile range. Statistical analysis between two groups was performed using the Mann–Whitney test.
Functional studies of BMP9 mutations
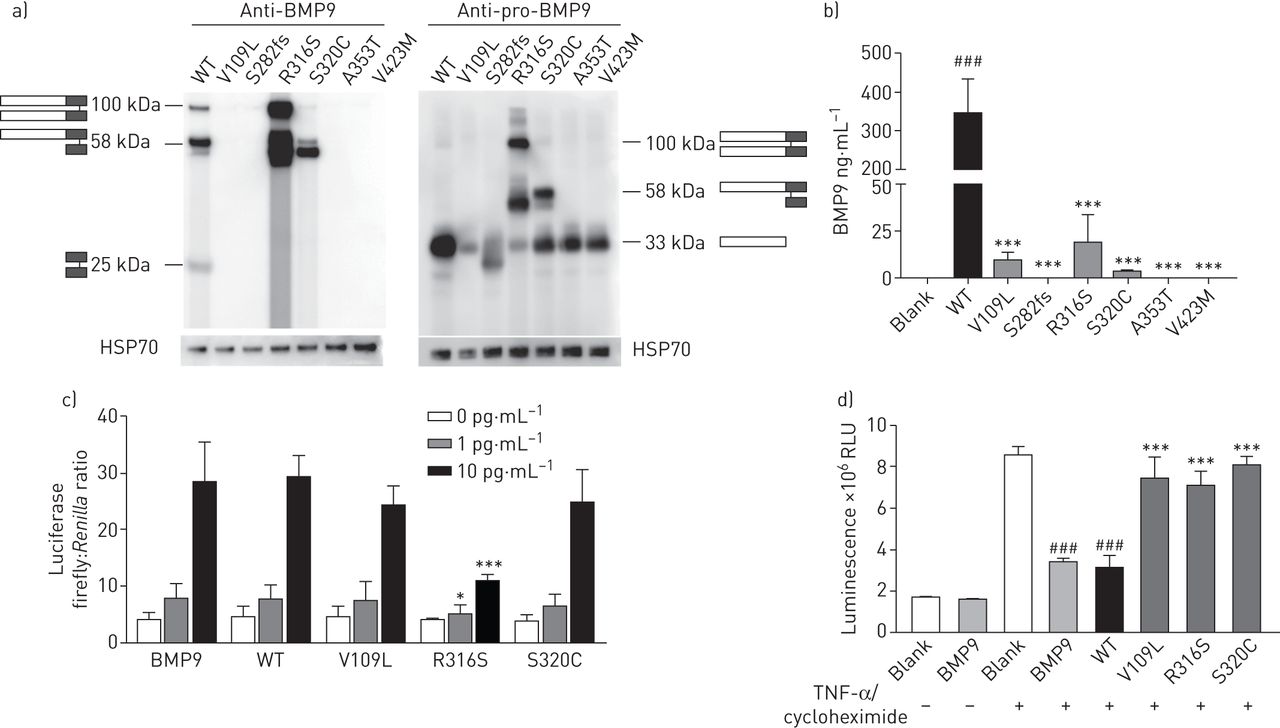
To investigate the deleterious outcomes resulting from BMP9 mutations, we selected six mutations for functional assessment based on the clinical presentations (supplementary figure S6). Three mutations (V109L, S282fs and V423M) were chosen because patients with these mutations had early onset of disease. The other three mutations (R316S, S320C and A353T) were studied because the mutation carriers had severe clinical manifestations that decreased in a short period of time after diagnosis (supplementary table S3). Western blot demonstrated that the wild-type (WT) and five missense plasmids (V109L, V423M, R316S, S320C and A353T) expressed similar levels of pro-BMP9 protein either in cell lysis (supplementary figure S7) or in supernatants from human embryonic kidney HEK293E cells (figure 4a, right panel). However, a significant decrease in mature BMP9 was observed for all BMP9 mutants (figure 4a, left panel). For the truncated mutation S282fs, neither pro-BMP9 nor mature BMP9 could be detected (figure 4a and supplementary figure S7). These results were confirmed by ELISA assay (figure 4b). The mature BMP9 level was significantly decreased in the culture supernatant of HEK293E cells transfected with V109L, R316S and S320C, and was almost undetectable for S282fs, A353T and V423M. We then used the conditioned media from V109L-, R316S- and S320C-transfected cells to examine their ability in activating the BMP signalling pathway (figure 4c). BRE (BMP response element)-luciferase activity assays revealed that R316S exhibited decreased activity compared with WT BMP9. Furthermore, pre-treatment of WT BMP9 conditioned media protected pulmonary arterial endothelial cells (PAECs) from apoptosis induced by tumour necrosis factor-α in the presence of cycloheximide. However, all of the three mutants were unable to prevent PAEC apoptosis (figure 4d).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Functional study of bone morphogenetic protein 9 (BMP9) mutations. WT: wild-type; HSP: heat shock protein; RLU: relative light units; TNF: tumour necrosis factor; PAEC: pulmonary arterial endothelial cell. a) Human embryonic kidney HEK293E cells transfected with mutant BMP9 showed a different expression pattern compared with WT. HEK293E cells were transfected with WT or BMP9 mutant plasmids. The supernatant was harvested for immunoblotting with an antibody specific to mature BMP9 or pro-BMP9. WT BMP9 protein fractionated as three bands corresponding to pro-BMP9 (100 kDa), partially processed pro-BMP9 (58 kDa) and mature BMP9 dimer (25 kDa). b) ELISA assay showed that the secretion of mature BMP9 was significantly lower in the supernatant of HEK293E cells transfected with mutant BMP9 than those from WT BMP9. ANOVA. ***: p<0.001 compared with WT; ###: p<0.001 compared with blank. c) The missense BMP9 mutation R316S caused significantly reduced BRE (BMP response element) activity compared with WT BMP9. After co-transfection with human activin receptor-like kinase 1, BRE-luciferase and pTK-Renilla, C2C12 cells were treated with WT or mutant BMP9 supernatant at 1 and 10 pg·mL−1, respectively. Commercial recombinant BMP9 protein was used as a positive control. ANOVA. *: p<0.05 compared with WT; ***: p<0.001 compared with WT. d) BMP9 mutations attenuated the anti-apoptosis effect of WT BMP9 in PAECs. PAECs were pre-treated with WT or mutant BMP9 supernatant at 5 ng·mL−1. The effect of the mutants of BMP9 on PAEC apoptosis was assessed by measuring caspase-3/7 activity following stimulation with TNF-α and cycloheximide. ANOVA. ***: p<0.001 compared with WT; ###: p<0.001 compared with blank.
Discussion
In the current study, we used NGS to identify that BMP9 is a novel IPAH susceptibility gene. The rare BMP9 mutations conferred a >21-fold greater odds of IPAH, ranking it second to BMPR2. Functional studies confirmed that these BMP9 mutations affected circulating BMP9 levels in IPAH patients. Furthermore, these mutants caused deleterious effects on protein expression and function in cultured cells. Taken together, these findings strongly suggest that mutations in BMP9 contribute to the aetiology of PAH.
NGS involving exome sequencing has led to the identification of novel genes for heritable PAH (HPAH) within families having multiple affected individuals [10, 11], However, challenges remain when applying this technology to identify novel genes among unrelated IPAH subjects. The difficulty stems from the recruitment of large homogeneous patient cohorts. In the current study, we recruited patients from two expert centres with consistent diagnostic procedures for IPAH. Almost all familial PAH (FPAH) patients carry a mutation of known PAH genes, and acute pulmonary vasoreactivity test-positive responders are different from IPAH patients with respect to the pathogenic mechanism and genetic aetiology [19]. Thus, we excluded these two subgroups of patients from the IPAH cohort to better decipher the genetic basis of IPAH. We enrolled 57 paediatric IPAH patients (mean±sd age 9.9±5.5 years), 274 adult patients (mean±sd age 31.8±7.7 years) and excluded cases >60 years old, resulting in a combined mean age of 28 years. In addition, all subjects participating in this study were officially registered as Chinese Han ethnicity. Therefore, the stringent selection criteria, precise clinical phenotype together with relatively young and homogeneous cohorts in the current study would generate convincing data on the IPAH-predisposing gene. As such, our data may also justify the higher mutation rate of the 10 previously identified PAH genes (38.7% versus 19.9%) and BMP9 (6.8% versus 1.1%) in the current study than reported in a European cohort by Gräf et al. [20].
The variations in genetic backgrounds among different populations and ethnicities result in the distinctive propensity of particular genes involved in rare diseases. For example, Factor V Leiden, an important risk factor in venous thromboembolism, has an average allele frequency of 4.4% in the European population, but is almost absent in the East Asian population [21]. Therefore, confirmation of putative genes involved in rare diseases between independent cohorts in different continents is crucial to identify the role of susceptible genes in the aetiology of the disease. Recently, Gräf et al. [20] performed WGS in European Caucasian IPAH/HPAH patients and reported a significant overrepresentation of mutations in four genes (ATP13A3 (ATPase 13A3), AQP1 (aquaporin-1), SOX17 (SRY-box 17) and BMP9). Among these genes, only BMP9 was validated in our study. None of the other three genes (ATP13A3, AQP1 and SOX17) achieved statistical significance in the combined cohorts (supplementary table S7). The mutations for the three genes are presented in supplementary dataset S2. Furthermore, among the 21 BMP9 mutations described in the current study, only two of the BMP9 mutations were reported previously [20]; the remaining 19 mutations were newly identified. Taken together, our results not only complement the previously reported European data by Gräf et al. [20], but also reinforce the critical role of BMP9 mutations in the genetic aetiology of IPAH.
BMP9 (also known as growth and differentiation factor 2) is predominantly expressed in the liver and constitutively secretes into the circulation. By binding to ACVRL1 (type 1 BMP receptor) and BMPR2 (type 2 BMP receptor), BMP9 plays an important role in regulating vascular biology and angiogenesis [18]. Because ACVRL1 and BMPR2 are highly expressed in pulmonary vascular endothelial cells, the optimum level of circulating BMP9 is essential to maintain the quiescence of the pulmonary vasculature [22]. Here, we provide compelling evidence indicating that BMP9 is a PAH susceptibility gene through an exome-wide study. All of the 21 BMP9 mutations identified are very rare, which is consistent with the low incidence of IPAH (six cases per million people per year). We have shown various BMP9 mutations could negatively affect BMP9 function, including synthesis, secretion, processing and/or activation of ACVRL1 signalling, which results in haploinsufficiency of a functional BMP axis in the pulmonary endothelium. Notably, the R316S mutation showed a unique protein expression pattern (figure 4a) and a more deleterious effect on ACVRL1 signalling (figure 4c). These results might be caused by the special location of the R316S mutation in the highly conserved cleavage site of BMP9, which could interfere with the proteolytic cleavage. The molecular mechanisms underlying the pathogenesis of PAH elicited by these BMP9 mutations deserve further investigation.
Of note, ACVRL1 ranked third among all the IPAH-related genes in our two cohorts, with rare mutations occurring in 22 cases. This was seemingly contradictory to the previous studies showing that ACVRL1 is a major disease-causing gene for HHT and PAH with HHT [23, 24]. To confirm our results, medical records and family histories were scrutinised to ensure that all these ACVRL1 mutation carriers were indeed IPAH. ACVRL1 mutation carriers might develop both PAH and HHT. In 78% patients carrying the ACVRL1 mutation from the French PAH Network, PAH diagnosis preceded manifestations of HHT [25]. Moreover, because HHT has nearly complete penetrance at the age of 60 years, some ACVRL1 mutation carriers might not exhibit clinical evidence of HHT when they are young. Given the younger age of the ACVRL1 mutation carriers identified in our study (mean (range) age 25.7 (3–50) years), these patients might not present the clinical features of HHT. We will keep following these patients to confirm the phenotype.
Recently, a subgroup of PAH patients was found to carry more than one rare gene variant, leading to a double heterozygous genotype. For example, Gräf et al. [20] reported that one PAH patient carried highly deleterious variants in both BMPR2 and SMAD9. In the current study, we identified eight cases carrying double mutations or rare variants in BMPR2/ACVRL1, BMPR2/BMPR1B, BMPR2/SMAD9, BMPR2/TBX4 and KCNK3/TBX4. The double heterozygosity for mutations or variants involving PAH risk genes highlights the complexity inherent to PAH. Whether the double variants contributed to PAH pathogenesis and clinical outcome warrants future study.
To the best of our knowledge, this is the first report to identify novel IPAH susceptibility genes in Asia using an exome-wide study based on a large population of IPAH patients. The identification of BMP9 mutations strongly supports the hypothesis that an impairment of genes involved in BMP signalling leads to PAH susceptibility. Experimentally, administration of recombinant BMP9 to stimulate BMP signalling in the endothelium can prevent or reverse PAH in several genetic and nongenetic rodent models [26]. Data in figure 3 indicate that circulating BMP9 levels decreased significantly in patients carrying BMP9 deleterious mutations. This finding suggests that IPAH patients carrying BMP9 mutants could receive BMP9 supplemental therapy as part of their personalised treatment.
Study limitations
A caveat of our study is that all the patients involved in the two cohorts were sporadic cases. Due to the unavailability of DNA samples from the affected relatives, familial segregation with PAH has not been demonstrated. Whether BMP9 mutations are associated with FPAH should be confirmed in pedigrees in the future. Another limitation is the relatively low number of patients in the replication cohort. Future studies among larger cohorts will be crucial to confirm further the association between BMP9 and IPAH. Given that the circulating BMP9 levels were only measured at baseline for the IPAH patients with or without mutations, we were unable to test the variability and prognostic value of BMP9 over time or the relevance to different therapeutic strategies.
Conclusions
Using NGS exome sequencing on large homogenous cohorts, we have confirmed that mutations in human BMP9 predispose to IPAH susceptibility.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material: Methods, tables and figures. ERJ-01609-2018_Supplement
Supplementary dataset 1: Rare variants and mutations in 10 previously reported PAH risk genes. ERJ-01609-2018_Dataset_1
Supplementary dataset 2: Rare variants in three candidate PAH genes. ERJ-01609-2018_Dataset_2
Acknowledgements
The authors gratefully thank the patients and families for their involvement in this study.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: X-J. Wang has nothing to disclose.
Conflict of interest: T-Y. Lian has nothing to disclose.
Conflict of interest: X. Jiang has nothing to disclose.
Conflict of interest: S-F. Liu has nothing to disclose.
Conflict of interest: S-Q. Li has nothing to disclose.
Conflict of interest: R. Jiang has nothing to disclose.
Conflict of interest: W-H. Wu has nothing to disclose.
Conflict of interest: J. Ye has nothing to disclose.
Conflict of interest: C-Y. Cheng has nothing to disclose.
Conflict of interest: Y. Du has nothing to disclose.
Conflict of interest: X-Q. Xu has nothing to disclose.
Conflict of interest: Y. Wu has nothing to disclose.
Conflict of interest: F-H. Peng has nothing to disclose.
Conflict of interest: K. Sun has nothing to disclose.
Conflict of interest: Y-M. Mao has nothing to disclose.
Conflict of interest: H. Yu has nothing to disclose.
Conflict of interest: C. Liang has nothing to disclose.
Conflict of interest: J.Y-J. Shyy has nothing to disclose.
Conflict of interest: S-Y. Zhang has nothing to disclose.
Conflict of interest: X. Zhang has nothing to disclose.
Conflict of interest: Z-C. Jing has nothing to disclose.
Support statement: The work was supported by grants from Beijing Natural Science Foundation (7181009, 7172180), National Key Research and Development Program of China (2016YFC0901502), National Natural Science Foundation of China (81320108005, 81630003, 81670052), National Science Fund for Distinguished Young Scholars (81425002), CAMS Innovation Fund for Medical Sciences (2016-I2M-1-002, 2016-I2M-4-003), and Director Fund from State Key Laboratory of Cardiovascular Disease (2017ZR-03, 2018ZR-02). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received August 24, 2018.
- Accepted November 29, 2018.
- Copyright ©ERS 2019
References










