



The use of whole-genome sequencing in cluster investigation of a multidrug-resistant tuberculosis outbreak
- Maeve K. Lalor1,2,
- Nicola Casali3,4,
- Timothy M. Walker5,
- Laura F. Anderson1,
- Jennifer A. Davidson1,
- Natasha Ratna1,
- Cathy Mullarkey6,
- Mike Gent7,
- Kirsty Foster8,
- Tim Brown3,
- John Magee9,10,
- Anne Barrett9,
- Derrick W. Crook5,11,
- Francis Drobniewski3,4,
- H. Lucy Thomas1 and
- Ibrahim Abubakar1,2
- 1Tuberculosis Section, National Infection Service, Public Health England, London, UK
- 2Institute for Global Health, University College London, London, UK
- 3PHE National Mycobacterium Reference Service South, Public Health England, London, UK
- 4Dept of Infectious Diseases, Imperial College London, London, UK
- 5Nuffield Dept of Medicine, John Radcliffe Hospital, University of Oxford, Oxford, UK
- 6TB Health Visiting Service, Leeds Community Healthcare, Leeds, UK
- 7Yorkshire and the Humber Public Health England Centre, Public Health England, Leeds, UK
- 8North East Public Health England Centre, Public Health England, Newcastle, UK
- 9PHE North of England Mycobacterium Reference Centre, Freeman Hospital, Newcastle, UK
- 10School of Biology, Newcastle University, Newcastle, UK
- 11National Infection Service, Public Health England, London, UK
- Maeve K. Lalor, National Infection Service, Public Health England, 61 Colindale Avenue, London, NW9 5EQ, UK. E-mail: maeve.lalor{at}phe.gov.uk
Abstract
We used whole-genome sequencing (WGS) to delineate transmission networks and investigate the benefits of WGS during cluster investigation.
We included clustered cases of multidrug-resistant (MDR) tuberculosis (TB)/extensively drug-resistant (XDR) TB linked by mycobacterial interspersed repetitive unit variable tandem repeat (MIRU-VNTR) strain typing or epidemiological information in the national cluster B1006, notified between 2007 and 2013 in the UK. We excluded from further investigation cases whose isolates differed by greater than 12 single nucleotide polymorphisms (SNPs). Data relating to patients' social networks were collected.
27 cases were investigated and 22 had WGS, eight of which (36%) were excluded as their isolates differed by more than 12 SNPs to other cases. 18 cases were ruled into the transmission network based on genomic and epidemiological information. Evidence of transmission was inconclusive in seven out of 18 cases (39%) in the transmission network following WGS and epidemiological investigation.
This investigation of a drug-resistant TB cluster illustrates the opportunities and limitations of WGS in understanding transmission in a setting with a high proportion of migrant cases. The use of WGS should be combined with classical epidemiological methods. However, not every cluster will be solvable, regardless of the quality of genomic data.
Abstract
Investigation of MDR-TB outbreaks with WGS was useful but it was often inconclusive whether transmission occurred http://ow.ly/Qk7130jPP5C
Introduction
Multidrug-resistant (MDR) tuberculosis (TB) is a major public health concern globally, with particularly high rates in Europe [1]. In the UK, the number and proportion of MDR-TB/rifampicin-resistant TB cases more than doubled from 38 cases (1.1%) in 2001 to 95 cases (1.8%) in 2011, although it has since decreased to 63 cases (1.7%) in 2016 [2]. A UK study of MDR-TB cases between 2004 and 2007, combining epidemiological information from cluster investigation with 24-loci mycobacterial interspersed repetitive unit variable tandem repeat (MIRU-VNTR) strain typing, estimated that up to 8.5% of UK MDR-TB cases arose from recent transmission [3].
Since January 2010, 24-loci MIRU-VNTR strain typing has been carried out on all culture-positive Mycobacterium tuberculosis complex isolates in the UK. Clusters of cases with indistinguishable strain types, which fulfil requisite criteria, have been investigated to inform public health action [4].
As well as providing diagnostic capability to identify M. tuberculosis complex and determine genotypic drug resistance, whole-genome sequencing (WGS) can determine the genetic relatedness between strains with greater resolution than MIRU-VNTR [5–8]. WGS has mainly been used retrospectively to assess transmission networks in outbreaks, including timing and direction of transmission, rather than prospectively during active cluster investigations [9–16]. In England, the roll-out of prospective WGS in routine TB diagnostics began in December 2016 and is expected to cover the whole of England by the end of 2017, replacing MIRU-VNTR [17].
In 2010, routine cluster review [4] led to the investigation of a possible TB outbreak of the Beijing strain (B1006; MIRU-VNTR: 424352332517333456443372) among UK residents. Of the 231 MDR-TB cases notified between 2010 and 2012, 62 (27%) clustered with at least one other MDR-TB case and 14 out of 62 (23%) were in B1006. As this was the largest MDR-TB cluster and was only the second in this time period to have more than five cases, it was considered to be of public health importance. The B1006 strain accounts for up to 25% of MDR-TB isolates tested in the former Soviet Union [18]. In November 2012, the status of the cluster investigation was raised to that of an incident, requiring a national level incident control team to be convened to consider what action to take [4]. This was due to an increase in the number of cases, including six with extensively drug-resistant (XDR) TB, and the suspicion that transmission of XDR-TB was occurring in the UK. As part of this intensified investigation, more detailed epidemiological information was collected and WGS was performed on isolates from patients.
Here we describe the impact WGS had on the on-going cluster investigation by ascertaining whether the greater discrimination of WGS reduced the number of cases initially identified by MIRU-VNTR that required further investigation, whether a clearer understanding of the transmission chain within this cluster was possible by combining the epidemiological links identified during the investigation and the WGS data and whether WGS helped to elucidate the direction and timing of transmission.
Methods
TB cases in the UK notified to the Enhanced Tuberculosis Surveillance System (ETS) were matched probabilistically [19] to laboratory results, including data on drug-susceptibility testing (DST) and MIRU-VNTR, from culture-positive isolates. The ETS collects demographic, clinical, social risk factor (SRF) and treatment outcome data. National TB clusters (defined as at least two cases with indistinguishable MIRU-VNTR strain types in more than one region) were identified using bespoke software and reviewed monthly in accordance with national guidance, with cluster investigations being launched where appropriate [4].
Cases of MDR-TB/XDR-TB notified in the UK between 2007 and 2013 with the B1006 MIRU-VNTR profile (424352332517333456443372), with at least 23 complete loci, were included and referred to as “B1006 clustered cases”. Culture-negative cases with a clinical diagnosis of TB and treatment for MDR-TB, which were epidemiologically linked to cluster B1006, were considered “probable clustered cases”. MDR-TB isolates with the B1006 strain from cases prior to 2010 that had been typed retrospectively were also included. Data relating to lifestyle and social networks were collected through questionnaires that case managers completed with their patients (see supplementary material).
A “confirmed epidemiological link” between two cases was defined as where either volunteered the name of the other as a contact, or where cases shared time in the same setting during the period when one of the cases was potentially infectious. A “probable epidemiological link” was defined where both cases had spent time in the same setting but the timing was uncertain. A “possible epidemiological link” was defined where two cases in the same geographical area shared social or behavioural traits (e.g. drug use) but a specific shared setting could not be established.
Cases were considered to still be clustered following WGS if sequencing data differed by 12 [9] or less single nucleotide polymorphisms (SNPs) from another B1006 case, or if they had an epidemiological link to a WGS clustered case. Several SNP cut-offs previously used in TB investigations (0–5 [11, 15] and 0–9) were explored as alternative thresholds to assess if they better captured transmission [15]. For methods on DNA preparation, WGS and phylogenetic analysis, refer to the supplementary material.
Results
Overall, 27 cases notified in the UK between 2007 and 2013 were included in cluster B1006, 24 of which were B1006 clustered cases and three were probable clustered cases (figure 1). The majority (20 out of 27) were non-UK born, including 14 who were born in Lithuania, with many (15 out of 27) having a history of SRFs.

Flow chart of cases in cluster B1006 (initial intensified investigation: n=27). 18 cases remained under investigation following whole-genome sequencing (WGS) results. These included: 14 clustered cases with less than 12 single nucleotide polymorphisms (SNPs) between each case and another case; three probable clustered cases with no culture (or WGS) which were epidemiologically linked to cases in the cluster; and one clustered case with mycobacterial interspersed repetitive unit variable tandem repeat (MIRU-VNTR) strain type B1006 but no WGS epidemiological link to another case in the cluster. MDR-TB: multidrug-resistant tuberculosis.
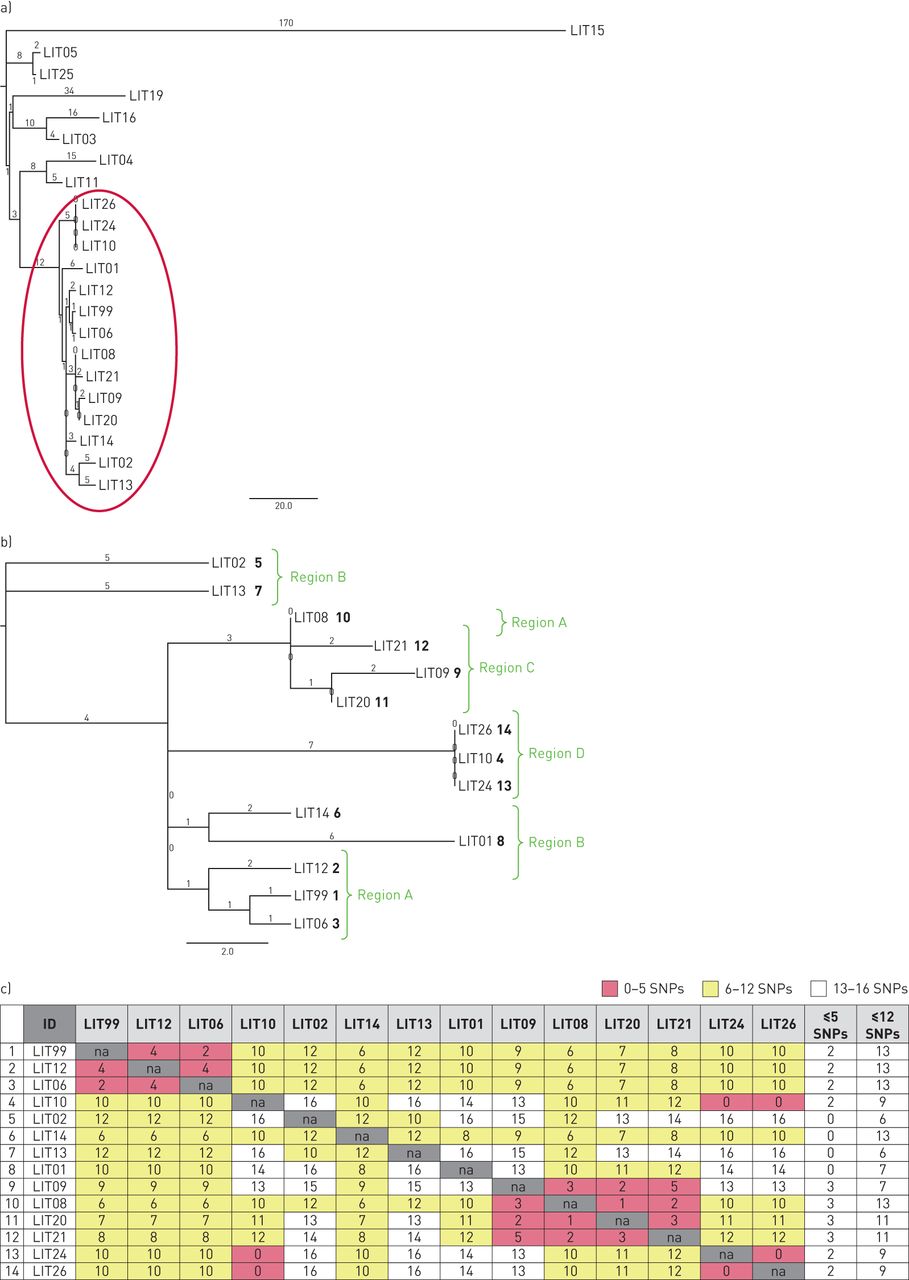
Some 22 patient isolates were sequenced and, of these, eight out of 22 isolates (36%) differed by more than 12 SNPs from all other isolates in the cluster (range of SNP difference: 30–179) (figure 2a). As none were epidemiologically linked within the cluster, they were excluded from further consideration. The remaining 14 cases were thus linked by WGS (figure 2b) and a further four with no sequencing data (three had no culture and one culture could not be regrown) were linked epidemiologically, resulting in a final cluster of 18 cases.

a) Phylogenetic tree of isolates showing genetic diversity between isolates sequenced from 22 cases in cluster B1006. The highlighted section shows isolates that have a maximum of 12 single nucleotide polymorphism (SNP) differences from at least one other isolate in the cluster; and b) phylogenetic tree of isolates sequenced from 14 cases with 12 or less SNP differences from at least one other isolate in the cluster. Numbers in regular text represent the number of SNPs while numbers in bold represent the order of cases in terms of symptom onset. Region labels indicate the geographical location of cases. c) SNP differences between isolates sequenced from 14 cases with 12 or less SNP differences from at least one other isolate in the cluster. The final two columns show how many cases the case is linked to using 5 or 12 SNP cut-offs.
Of these 18 cases, 13 (72%) had at least one SRF (i.e. drug misuse (n=6), alcohol misuse (n=5), homelessness (n=5), imprisonment (n=9), or previous TB treatment (n=4)). 11 out of 18 cases were born in Lithuania, six in the UK and one in South-East Europe. Three were children under the age of 5 years (all born in the UK, two to Lithuanian parents). 12 out of 18 cases (66%) were notified between August 2012 and August 2013, including five UK born patients, suggesting possible transmission within the UK (figure 3). 15 out of 18 cases (83%) had pulmonary disease, 11 of whom (61%) were sputum smear positive. Five of these pulmonary cases were symptomatic for at least 6 months before starting treatment, one of whom (LIT12) was symptomatic for 2 years, while all five had chaotic lifestyles with multiple SRFs. Two cases (LIT13 and LIT24) remained culture positive for more than a year after starting treatment.

Timeline from onset of symptoms until the start of multidrug-resistant tuberculosis (MDR-TB) treatment for cases in B1006, showing their characteristics. XDR-TB: extensively drug-resistant tuberculosis.
All 15 culture-confirmed cases in the cluster had isolates with phenotypic resistance to isoniazid, rifampicin, ethambutol, streptomycin and kanamycin. There were differences in DST for pyrazinamide, prothionomide, ethionomide, moxifloxacin and ofloxacin (figure 3). The eight cases in Region A and Region C shared the same DST profile and had pyrazinamide resistance; the three cases in Region D shared the same profile and were sensitive to pyrazinamide; and the four XDR-TB cases in Region B had resistance to moxifloxacin and ofloxacin, three of whom were also resistant to prothionamide and ethionamide.
Potential transmission networks
Amongst the 14 cases with isolates within 12 SNPs of each other, each was genomically linked to between six and 13 people; however, there was clear sub-clustering within each geographic region (figures 2b and 2c). Using the five SNP threshold suggested there were three unlinked local outbreaks (figure 4a). Increasing the threshold to nine SNPs (figure 4b) suggested transmission may have been more widespread between Region A, Region B and Region C, and increasing the threshold to 12 SNPs suggested transmission may have occurred across all geographical areas (figure 2c). By combining the epidemiological information, DST profiles and genomic data, potential transmission networks were identified in four regions of England (figure 5).

Cluster diagram showing epidemiological links and whole-genome sequencing (WGS) data nationally, with (a) five or less single nucleotide polymorphisms (SNPs) or (b) nine or less SNPs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cluster diagram showing epidemiological links and whole-genome sequencing (WGS) data within four areas (Regions A–D, represented by (a), (b), (c) and (d), respectively). Cases who spent time in more than one geographical area are presented in each region.
In Region A (figure 5a), the first case (LIT 99) was UK born, had extrapulmonary TB and no SRFs. She shared no distinctive characteristics with, or epidemiological links to, the other cases in Region A, despite her isolate being two and four SNPs from those of two Lithuanian cases. The low number of SNPs is consistent with presumed recent transmission, however, given that the first case had extrapulmonary TB and there was no supporting epidemiological evidence it is unlikely she was the source of infection for the subsequent cases and it was therefore inconclusive as to whether recent transmission had occurred. There was one known epidemiological link between two prisoners, but only one had WGS results. The three Lithuanian cases whose isolates were separated by 4–6 SNPs had all worked in the construction industry, although it is unknown whether they had ever encountered each other. All cases had the same phenotypic DST profile.
In Region B (figure 5b) there was one probable epidemiological link between two cases who had lived together at some point (timing and location unknown). However, their isolates had a 12 SNP difference and different DST profiles, making direct transmission unlikely. All four XDR-TB cases in this location worked in the agricultural industry for agencies and had frequent job changes. Three of them reported smoking cannabis and three had been in prison in Lithuania, although at different times to each other. The XDR-TB cases had all previously been treated for TB, three on more than one occasion in Lithuania (strain type and sensitivities of initial treatment unknown). The four XDR-TB cases had between eight and 12 SNPs difference between their isolates and two had different resistance profiles. No confirmed epidemiological links between them could be identified. Evidence for transmission of XDR-TB was therefore inconclusive as the epidemiological, microbiological and genomic data were not supportive when combined.
In Region C (figure 5c), the first four cases notified were within two families who shared a house. Another case was linked to the workplace where the adults in the household worked and a further UK born case was treated in the same hospital that the other cases were treated at, but no definitive setting or timing was identified. All cases with WGS were five or fewer SNPs apart from at least one other case, confirming the epidemiological evidence for transmission. The most parsimonious interpretation of the phylogenetic tree suggested LIT08 may have transmitted to LIT20 and LIT21 and that LIT20 may have transmitted to LIT09 (figures 2a and 2b). However, closer inspection of the sequence data suggests LIT08 is more likely to have been the source for both LIT20 and LIT09, as the variant distinguishing these two sequences from LIT08 is present as a minority allele in LIT08. The epidemiological information did not support LIT20 transmitting to LIT09 but supported the interpretation of the variants that LIT09 was likely to have been infected by LIT08 (the child's parent).
In Region D (figure 5d), the first two cases notified lived close to each other and had indistinguishable isolates. However, despite intensive investigations, no common setting or evidence that they knew each other was identified. General awareness-raising regarding the signs and symptoms of TB was undertaken with providers of services for vulnerable groups and information about local TB services was provided; however, no active case finding was undertaken. Two further cases were household contacts of the first and second cases and the isolate of the household contact of the first case was indistinguishable to that of the first case itself. No sample was taken from the other household contact. The WGS results supported the epidemiological data that transmission had occurred. The direction of transmission was inferred from the epidemiological data, but as there were zero SNP differences between isolates, no additional information relating to timing or the direction of transmission could be determined by the WGS results.
Identification of epidemiological links and transmission settings: added value of WGS
A total of 24 epidemiological links between cases were identified (table 1), nine (38%) through routine contact tracing (seven household contacts, one relative contact and one work contact) and an additional 15 epidemiological links following MIRU-VNTR cluster investigation (one prison, one work, one hospital, two neighbour and one probable previous household contact, as well as nine possible work/drug use contacts). Two of the nine links that were identified through routine contact tracing and five out of 15 links identified following MIRU-VNTR results were confirmed by WGS. Of the remaining 10 links identified through MIRU-VNTR investigation, seven had six to 12 SNPs between them and were thus unclear, two had more than 12 SNPs and were thus refuted by WGS and one could not be re-grown for WGS. Two new settings were identified, a workplace and a hospital, and public health actions were undertaken at the workplace but no new cases were identified.
Epidemiological links identified following the action taken (contact tracing, mycobacterial interspersed repetitive unit variable tandem repeat (MIRU-VNTR) cluster investigation and whole-genome sequencing (WGS))
Discussion
A large MDR-TB/XDR-TB cluster was identified in the UK by MIRU-VNTR strain typing, enabling prompt cluster investigation. Detailed analyses of epidemiological and genomic data provided strong evidence that transmission had occurred in the UK. Consistent with the recognised limitations of MIRU-VNTR for the Beijing lineage [20], the use of WGS allowed discrimination between cases clustered by MIRU-VNTR and the exclusion of one-third of cases from the investigation. This allowed resources to be focused on the investigation of cases that were more likely to have been part of the same transmission network.
As well as the evidence WGS provided to refute transmission that MIRU-VNTR had identified, WGS also provided corroborative evidence that transmission was likely to have occurred in a small number of cases which had either confirmed or probable epidemiological links. In most cases the WGS data were consistent with the combined findings from MIRU-VNTR and epidemiological investigation. In one instance WGS suggested transmission could have occurred (separated by two and four SNPs to other cases), yet this seemed epidemiologically implausible as the case with earliest symptom onset had extrapulmonary TB and no known links with two subsequent cases. One possible explanation for this may be the presence of unknown intermediary cases.
Whilst there are anecdotal data where phylogeny has indicated the direction of transmission in TB outbreaks [15], this analysis also underscores the importance of exploring the raw sequence data at variant sites. During the active investigation of this cluster, as well as in the routine use of WGS data in cluster investigation, only the phylogenetic tree was used to direct public health action. Only after the incident, when trying to understand why the tree and epidemiological data were inconsistent, did we find the presence of a variant allele, seen in secondary strains, as a minority allele in an ancestral strain. This deeper analysis of the sequence data was in concordance with the epidemiological data and aided in understanding the possible direction of transmission between cases in this cluster, as was recently demonstrated by Worby et al. [21] in other pathogens. However, the addition of WGS did not facilitate the identification of additional links or missing cases identified in this setting.
The most effective SNP threshold to apply in practice, to most efficiently identify transmission networks in TB, has been discussed previously [16–18, 22]. While our results suggest that identifying large numbers of SNPs between isolates is extremely helpful for refuting transmission, we found that the 12 SNP cut-off is likely to over-estimate recent transmission. For example, significant inter-regional transmission in this cluster didn't look plausible and small SNP differences between isolates did not always lead to the identification of epidemiological links, as was also recently shown in a large isoniazid cluster in London [22]. Although distances of 0–2 SNPs, or even 0–5 SNPs, would normally suggest a high probability of recent transmission, this outbreak highlights the need to remain aware of exceptions to this, particularly when a large number of cases occur in migrants whose disease may be due to reactivation of distantly acquired infection. Other factors may also affect the assessment of transmission through SNP differences, such as clinical disease manifestation, duration of infection, patient bacterial load, antibiotic therapy, acquisition of drug resistance and actual infective dose, which would need building into any model using SNP number to predict transmission events.
Many of the patients in this cluster had chaotic lifestyles and multiple risk factors associated with delayed diagnosis and poor adherence to treatment, plausibly contributing to further transmission of this strain. Cluster investigation in populations with a high number of risk factors and drug-resistant TB requires considerable resources. The lifestyles of some of the cases in this cluster presented a challenge to TB control, as it was difficult to collect epidemiological information. Indeed, epidemiological links between cases who were genotypically linked may not have been identified in this cluster. While such investigations can be resource intensive, the prevention of TB transmission in high risk groups, including of drug-resistant TB, means cluster investigation remains a key component of disease control.
Routine cluster investigation based on MIRU-VNTR was recently scaled back in the UK, in part due to a lack of evidence on its cost-effectiveness in preventing further transmission and in part due to a lack of available resources [23]. Considerable time and resources were frequently invested without identifying additional epidemiological links, transmission settings, or cases. This may be due in part to MIRU-VNTR not being able to adequately distinguish clusters in some TB lineages or due to the high proportion of imported TB cases which reactivate in the UK [24]. Other low-incidence settings have found similar issues relating to the specificity of MIRU-VNTR [25]. To address this concern, a more targeted approach for flagging high risk national MIRU-VNTR clusters for review, by running a red flag algorithm to identify priority clusters (including clusters with cases with MDR-TB), was developed in England. Furthermore, due to the poor resolution of MIRU-VNTR for some TB lineages [20] and high levels of clustering in non-UK born patients with no identified epidemiological links, many clusters in the UK with predominantly non-UK born populations have been assumed to represent common imported strain types and investigation has not been prioritised. The results from this analysis show transmission may be occurring in the UK in a subset of these clusters. Recent analysis suggests WGS is cost-effective due to the parallel identification of drug-resistant strains [5]. Due to the ability of WGS to predict drug resistance and its greater resolution, replacement of MIRU-VNTR strain typing may be cost-effective. Modelling the cost·(quality adjusted life year)−1 gained with this technology would be useful.
WGS is now being used routinely in the TB diagnostic pathway in England [26]. Potential benefits will include faster results for speciation of mycobacteria, as well as prediction of drug susceptibility and relatedness of cases in a single process [5]. The long term systematic use of WGS should also enable better analysis of transmission dynamics at a population level in England than was possible with MIRU-VNTR, in order to monitor the impact of policy changes on transmission [27, 28]. As the roll-out of WGS is underway, both clinical and public health teams have begun to make use of this new technology and have described both its benefits (in an XDR-TB cluster in London) and its limitations (in a large isoniazid-resistant cluster in London) [22, 29].
Bayesian inference methods, which combine information on SNP differences between isolates, time to nearest common ancestor and epidemiological data, are likely to be of benefit for helping to understand possible transmission networks and to inform public health action [30]. If automated algorithms can be applied combining WGS data and epidemiological data, informed by highly predictive models, then cluster investigation may be considered in middle-income settings as well as other high-income settings. In resource-poor settings, where TB transmission is likely to be more common, little use has been made of real-time cluster investigation largely due to lack of available resources.
The introduction of universal WGS in England will undoubtedly revolutionise testing for antibiotic resistance [26]. In addition, using WGS in this cluster investigation provided evidence that a third of the cases identified on the basis of MIRU-VNTR type were not plausibly part of the same transmission network, thus enabling us to focus additional investigative resources on a smaller number of cases. WGS also provided supportive evidence that transmission had occurred in a small number of cases with confirmed epidemiological links. Despite the obvious increase in granularity of WGS data, the evidence on whether some cases in this cluster were part of the transmission chain was inconclusive, especially in non-UK born cases. These data suggest that, as was seen using MIRU-VNTR, WGS in combination with epidemiological investigation may not enable the determination of whether recent and hence UK-based transmission has occurred in all cases. This study emphasises the importance of the use of classical epidemiological methods, as a significant proportion of the epidemiological links we identified were as a result of routine contact tracing. The use of genotyping data alone, whether that be MIRU-VNTR or WGS, likely over-estimates transmission resulting in inconclusive determination of networks. Whilst WGS is best viewed as a tool that directs epidemiological investigations with optimal precision, future research should evaluate the impact of WGS use on subsequent public health action and detection of previously unrecognised cases in cluster investigation. This may allow the re-initiation of routine prospective cluster investigations in resource-rich, low-incidence settings approaching TB elimination.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material (appendix) ERJ-02313-2017_Supplement
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: M.K. Lalor, H.L. Thomas and I. Abubakar conceived and designed the study. M.K. Lalor, L.F. Anderson, J.A. Davidson, N. Ratna, C. Mullarkey, M. Gent, K. Foster and F. Drobniewski collected data for the study. T.M. Walker and N. Casali conducted sequencing and sequencing analysis for the study. M.K. Lalor carried out the analysis and wrote the manuscript. T. Brown, J. Magee, A. Barrett and F. Drobniewski provided isolates for the study. All authors were involved in the incident, contributed to the design of the analysis and commented on manuscript versions. All authors have read and approved the final version of this manuscript. Ethics approval and consent to participate: Public Health England has authority under the Health and Social Care Act 2012 to hold and analyse national surveillance data for public health and research purposes.
Conflict of interest: None declared.
This article has supplementary material available from erj.ersjournals.com
Support statement: This study was supported by Public Health England, no external funding was received. F. Drobniewski and N. Casali were supported by the Imperial Biomedical Research Centre.
- Received November 9, 2017.
- Accepted April 26, 2018.
- Copyright ©ERS 2018