





Abstract
Novel anti-inflammatory approaches targeting chronically activated kinase pathways in chronic obstructive pulmonary disease (COPD) are needed. We evaluated RV568, a p38 mitogen-activated protein kinase-α and -γ and SRC family kinase inhibitor, in cellular and in vivo models relevant to COPD and examined its safety and efficacy in COPD patients.
The anti-inflammatory activities of RV568 were tested in primary cultured monocytes, macrophages and bronchial epithelial cells and in vivo in lipopolysaccharide and cigarette smoke-exposed murine models. RV568 was evaluated in a 14-day trial in COPD patients.
RV568 showed potent anti-inflammatory effects in monocytes and macrophages, which were often greater than those of corticosteroids or the p38 inhibitor Birb796. RV568 combined with corticosteroid had anti-inflammatory effects suggestive of a synergistic interaction in poly I:C-stimulated BEAS-2B cells and in the cigarette smoke model. In COPD patients, inhaled RV568 (50 µg and 100 µg) improved pre-bronchodilator forced expiratory volume in 1 s (69 mL and 48 mL respectively) and significantly reduced sputum malondialdehyde (p<0.05) compared to placebo, although there were no changes in sputum cell counts. Adverse events during RV568 and placebo treatment were similar.
RV568 shows potent anti-inflammatory effects on cell and animal models relevant to COPD. RV568 was well-tolerated and demonstrated a modest clinical benefit in a 14-day COPD clinical trial.
Abstract
Kinase inhibitor RV568 is anti-inflammatory in COPD models and shows signs of clinical efficacy in a small trial http://ow.ly/aASJ30fjOQL
Introduction
Chronic obstructive pulmonary disease (COPD) is characterised by excessive pulmonary inflammation in response to exposure to noxious particles, commonly from cigarette smoking [1]. Inhaled corticosteroids (ICS) are the most commonly used anti-inflammatory drug in COPD, but these drugs have limited clinical benefits and can cause side-effects [2]. There is a need for novel anti-inflammatory approaches to treating COPD.
Airway inflammation in COPD involves different inflammatory cell types, including neutrophils, macrophages, lymphocytes and epithelial cells [3]. A range of intracellular kinases regulate the inflammatory responses of these cells. p38 mitogen-activated protein kinase (p38 MAPK) is involved in inflammatory cytokine transcription, mRNA stabilisation and protein translation [4, 5]. p38 MAPK activation is increased in the lungs of COPD patients, and p38 MAPK inhibitors suppress cytokine secretion from COPD alveolar macrophages, lymphocytes and bronchial epithelial cells [6–9]. However, p38 MAPK inhibitors have shown variable effects in COPD clinical trials, with some long-term studies showing no clinical benefit [10–12]. There are four p38 MAPK isoforms: α, β, γ and δ. p38 MAPK inhibitors with α isoform selectivity have been used for clinical trials, as this isoform is expressed on immune cells. The γ isoform is expressed in COPD lung tissue and plays a role in corticosteroid-resistant inflammation [13]. Targeting of the p38 MAPK-α isoform alone will not suppress pro-inflammatory signalling by other p38 MAPK isoforms, or by other kinase pathways involved in the pathophysiology of COPD [14].
A series of small-molecule agents that inhibit a selected set of kinases involved in COPD inflammation has recently been developed, including RV568 [15]. These so-called “narrow-spectrum kinase inhibitors (NSKIs)” were designed for high potency against both the α and γ isoforms of p38 MAPK. Furthermore, chemical modifications ensured activity against the SRC kinase family, specifically haematopoietic kinase (HCK). HCK is involved in the macrophage pro-inflammatory response to lipopolysaccharide (LPS) stimulation, and is overexpressed in peripheral blood mononuclear cells (PBMCs) from COPD patients [16, 17]. These NSKIs are designed for inhaled delivery with prolonged lung retention in order to facilitate once-daily dosing and to minimise the potential for systemic side-effects. Furthermore, p38 MAPK inhibition may increase the anti-inflammatory effects of corticosteroids, by mechanisms such as enhanced glucocorticoid receptor translocation [18]. A potential attribute of NSKIs such as RV568 may be synergistic pharmacological activity with corticosteroids.
We have characterised the anti-inflammatory activity of RV568 using in vitro and in vivo models relevant to COPD inflammation. The effects of RV568 were compared with those of corticosteroids, and we investigated the pharmacological effects when used in combination with a corticosteroid. We also report a randomised, placebo-controlled clinical trial of inhaled RV568 in COPD patients, investigating principally the safety of this novel drug, but also providing an opportunity to study anti-inflammatory effects.
Materials and methods
Cellular assays
Monocytic and macrophage-like cells were stimulated with the bacterial protein LPS; THP1 cells, PBMCs, differentiated THP1, phorbol 12-myristate 13-acetate-differentiated U937 (d-U937) cells and sputum macrophages were used. The Toll-like receptor 3 (TLR3) agonist poly I:C and cytokine tumour necrosis factor-α (TNF-α) were used to stimulate the following epithelial cells: primary bronchial epithelial cells from healthy subjects and BEAS-2B cells. The effects of RV568, Birb796 (a pan-p38 inhibitor) and the corticosteroid fluticasone propionate (FP) on cytokine release and intercellular adhesion molecule (ICAM)-1 protein expression were measured. Full details of the methods can be found in the online supplementary material.
Mouse models
A murine model of inhaled LPS-induced pulmonary inflammation and a murine cigarette smoke model of pulmonary inflammation were used as previously described [15]. Bronchoalveolar lavage (BAL) was performed to measure cell counts and inflammatory proteins. Further details are in the online data supplement.
Clinical trial
A randomised, double-blind, parallel-group study (clinical trials.gov NCT01475292) was conducted at the Medicines Evaluation Unit (Manchester, UK). 30 COPD patients were recruited. Subjects were current or ex-smokers, aged 40–75 years, with a post-salbutamol forced expiratory volume in 1 s (FEV1) ≥40% and ≤80% predicted and an FEV1/forced vital capacity ratio ≤0.70. Patients with significant other medical conditions were excluded. Participants were allowed to remain on their current inhaled treatment throughout the study. The use of systemic corticosteroids (oral, parenteral or depot), oral beta-2 agonists, theophyllines, phosphodiesterase inhibitors, oral leukotriene inhibitors or antibiotic therapies was prohibited throughout the study. Subjects were randomised (1:1:1 ratio) to receive RV568 50 µg, RV568 100 µg or placebo once daily for 14 days.
All delivered doses were administered using a Philips Respironics I-neb Adaptive Aerosol Delivery (AAD) system (Philips Respironics, Amsterdam, The Netherlands). The following pre-defined criteria were used to withdraw subjects during the treatment period to ensure safety: 1) exacerbations requiring treatment with oral corticosteroids and/or antibiotics, 2) a decrease in post-bronchodilator FEV1 >20%, and 3) FEV1 <35% predicted. The primary end-point was safety, as assessed by: adverse events, ECG parameters, clinical laboratory evaluations (haematology, clinical chemistry and urinalysis) and FEV1. Other end-points included pharmacokinetic (PK) parameters and sputum biomarker analysis for treatment effects (immunoassays are described in the online supplementary material).
Statistical analysis
The sample size for the clinical study was based on safety considerations, to expose a limited number of COPD patients to a novel drug. Safety analyses included all subjects who received ≥1 dose. PK analyses included all subjects with ≥1 PK sample. FEV1 and sputum biomarker analyses were conducted using analysis of covariance. Analyses of in vitro and in vivo experiments were performed by either ANOVA or t-test. The IC50 (median inhibitory concentration) values were calculated using GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA) or XLfit software package (IDBS, Guildford, UK). The combination index for the interaction between two drugs was performed as previously described [19].
Results
Effects of RV568 in monocyte and macrophage cells
LPS-induced CXCL8 release was measured in PBMCs from healthy volunteers. RV568 or Birb796 exerted concentration-dependent CXCL8 inhibition (fig. 1a). The Emax (maximal response) for RV568 (81%) was greater than that for Birb796 (60%) and FP (50%). The results of similar experiments using THP1 cells and COPD PBMCs are described in the online supplement (online supplementary table S1).

RV568 reduces inflammation in monocytes, macrophages and epithelial cells. a) Inhibition of lipopolysaccharide (LPS)-induced CXCL8 release in healthy peripheral blood mononuclear cells (PBMCs). PBMCs were isolated from whole blood of healthy volunteers (n=3) and treated with 0.1 μg·mL−1 LPS for 4 h. b) Inhibition of LPS-induced CXCL8 release in d-U937 cells. U937 cells were differentiated into a macrophage-like cell with phorbol 12-myristate 13-acetate (n=3) and treated with 0.1 μg·mL−1 LPS for 4 h. c) Inhibition of tumour necrosis factor (TNF)-α-induced interleukin (IL)-6 release in normal human bronchial epithelial cells (NHBEs). NHBEs (n=3) were stimulated with 50 ng·mL−1 TNF-α for 4 h. d) Inhibition of TNFα-induced CXCL8 release in NHBEs. NHBEs (n=3) were stimulated with 50 ng·mL−1 TNF-α for 4 h. Cytokine releases were measured in the cell-free supernatants by ELISA; the per cent inhibitions were calculated by comparison with vehicle controls. Data are presented as mean±sem; ANOVA with Dunn's test, p<0.05 to RV568.
CXCL8 release in LPS-stimulated d-U937 cells was examined (fig. 1b). Birb796 had little effect on CXCL8, while RV568 and FP both showed concentration-dependent CXCL8 inhibition, with RV568 reaching higher Emax values (95%) than FP (80%), while FP demonstrated greater potency with >100-fold lower IC50 values (92 nM versus 0.006 nM). The results of other experiments using d-U937 and differentiated THP1 cells are described in the online supplement (online supplementary table S1). Only RV568 significantly inhibited CXCL8 release from COPD sputum macrophages (55% inhibition; online supplementary figure S1b, online supplementary table S3).
Inhibition of cytokine release in epithelial cell systems by RV568
Using primary normal bronchial epithelial cells, we examined TNFα-induced interleukin (IL)-6 and CXCL8 release; RV568 inhibited cytokine release in a concentration-dependent manner, with Birb796 and FP demonstrating lower effects (fig. 1c and d). Stimulation of BEAS-2B cells with poly I:C increased ICAM-1 protein expression. Again, this was inhibited by RV568 (Emax 97%), while Birb796 and FP had no effect (online supplementary figure S2).
The effects of combining dexamethasone (Dex) and RV568 on the release of IL-6 and CXCL8 were examined in poly I:C-stimulated BEAS-2B cells (fig. 2 and online supplementary figure S3). Both drugs alone showed concentration-dependent cytokine inhibition. Dexamethasone with 10 nM RV568 caused greater inhibition of cytokine release than dexamethasone alone. Further increases in the concentration of RV568 in combination with the corticosteroid produced progressively greater inhibition of cytokine production. Many of these combination conditions demonstrated synergism when analysed using the combination index (online supplementary table S4).
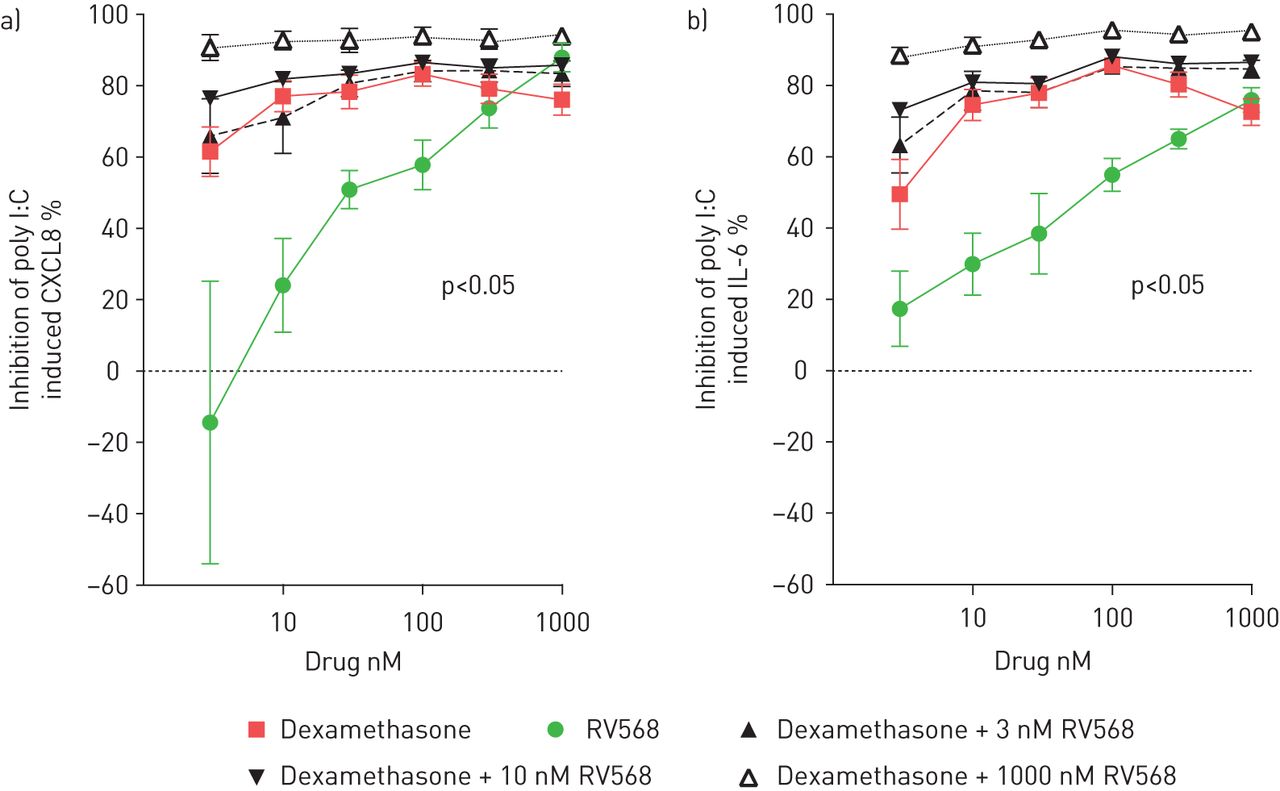
Inhibition of poly I:C-induced cytokine release in BEAS-2B cells. Cells were pre-incubated with dexamethasone alone, RV568 alone and both drugs combined at all indicated concentrations for 1 h prior to stimulation with poly I:C. a) Inhibition of poly I:C-induced CXCL8. b) Inhibition of poly I:C-induced interleukin (IL)-6. Cytokine release was measured in the cell-free supernatants by ELISA; the per cent inhibitions were calculated by comparison with vehicle control. Data are presented as mean±sem; n=8; ANOVA with Dunn's test, p<0.05 to RV568.
In vivo profiling of RV568
The murine LPS model was used to evaluate RV568 for potency and duration of action. RV568 was delivered intratracheally in a solution formulation 2 h prior to LPS inhalation. RV568 prevented the accumulation of neutrophils in the BAL fluid (online supplementary figure S4). The experiment was repeated using a suspension formulation, and RV568 showed similar effects (fig. 3a). Furthermore, RV568 significantly inhibited neutrophil accumulation when administered up to 8 h prior to LPS inhalation (fig. 3b).

RV568 inhibits lipopolysaccharide (LPS)-induced neutrophilia with a long duration of action in the mouse. a) Mice (n=8) were intratracheally administered a solution of vehicle or RV568 2 h prior to LPS exposure via inhalation. b) Mice (n=8) were intratracheally administered a solution of vehicle or RV568 at the indicated time points prior to LPS exposure. Bronchoalveolar lavage was collected 8 h after the LPS challenge and neutrophil influx measured. Data are presented as mean±sem; ANOVA with Dunnett's test, *: p<0.05 to vehicle control.
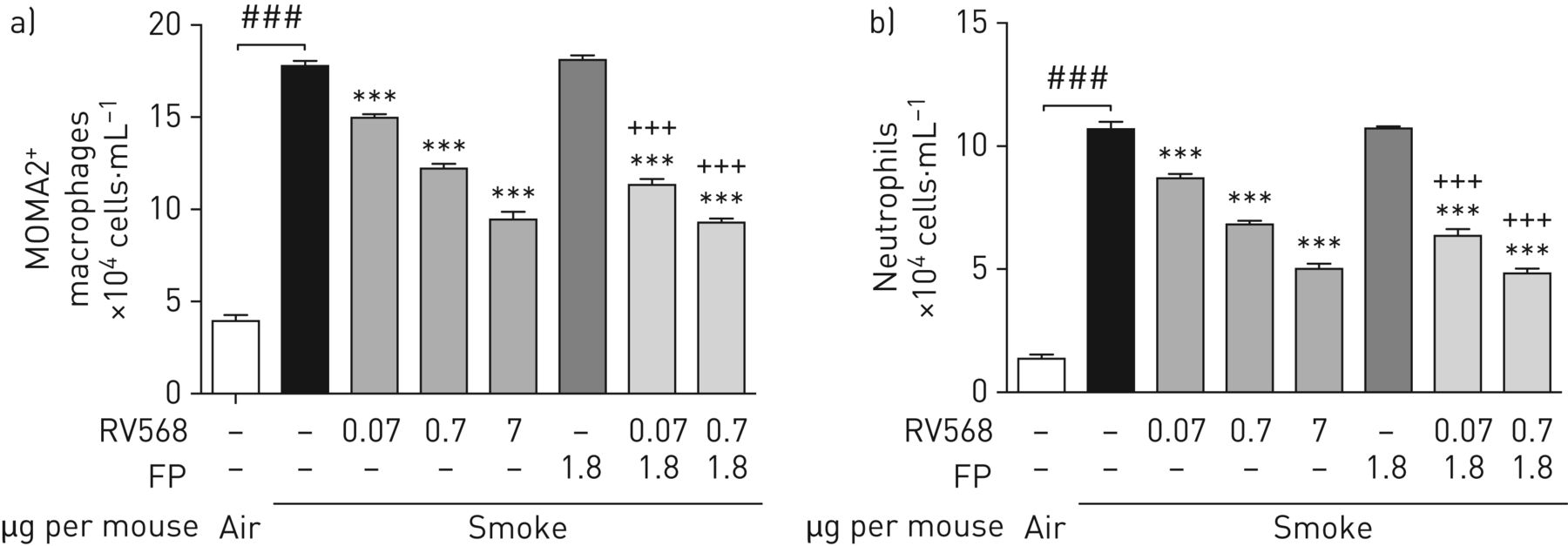
The therapeutic effects of RV568 administered to mice after 11 days of cigarette smoke exposure were examined. Figure 4 shows that fluticasone propionate failed to reduce BAL macrophage and neutrophil numbers on day 15. RV568 reduced these cell numbers in a dose-dependent manner. When RV568 was combined with the ineffective FP dose, the effects were significantly potentiated compared to the effect with RV568 alone. Inflammatory mediator levels were measured in BAL supernatants. RV568 significantly reduced chemotactic mediators, Th1 and Th17 cytokines, remodelling markers as well as the oxidative stress marker malondialdehyde (MDA) (online supplementary table S5). In all circumstances, the combination of RV568 and FP resulted in significant differences from RV568 or FP alone.

Therapeutic administration of RV568 attenuates smoking-induced inflammation in the mouse. Mice (n=5–6 per group) were exposed to 30 min of cigarette smoke once daily for 11 days followed by 3 days of once-daily intranasal administration of vehicle, fluticasone propionate (FP), RV568 or indicated drug combinations (μg per mouse). Bronchoalveolar lavage (BAL) was collected 24 h after the last drug administration and the numbers of alveolar macrophages and neutrophils were determined by fluorescence-activated cell sorting (FACS) analysis. a) MOMA2+-macrophage accumulation in BAL. b) Neutrophil accumulation in BAL. Data are presented as mean±sem; ANOVA with Dunnett's test: ***: significant difference from cigarette smoke control at p<0.001. ###: significant difference versus air control at p<0.001 and +++: significant difference versus FP treatment at p<0.001, by t-test with Welch's correction.
Clinical trial of RV568 in COPD patients
There were 71 screen failures, mainly due to the presence of other significant medical conditions (online supplementary table S2). The demography of the 30 participants is shown in table 1. The mean FEV1 and smoking status (current or former) and inhaled medication use were similar between groups, although more patients treated with placebo group used triple therapy (two long-acting bronchodilators plus inhaled corticosteroid).
COPD patient demographics
Two patients were withdrawn from the study due to a fall in FEV1>20% from the baseline value; one was withdrawn after a single RV568 100 μg dose with no accompanying symptom changes, the other after 6 days treatment with RV568 50 μg with flu-like symptoms. The adverse event profile was similar between treatment groups (online supplementary table S6). There were no significant ECG changes in any subjects.
There were significant improvements in the pre-bronchodilator FEV1 after 14 days’ treatment with RV568 100 μg and 50 μg compared to placebo (mean treatment differences of 48 mL (p=0.014) and 69 mL (p=0.0067), respectively (fig. 5a)), with similar effects in patients using and not using ICS. There was an improvement in residual volume with RV568 100 μg compared to placebo (median changes at 14 days compared to baseline were −0.06 L and 0.125 L, respectively, p=0.038; online supplementary figure S5). There were no significant changes in other lung volume measurements or specific airway conductance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical and sputum biomarker improvements after a 14-day administration of RV568 in chronic obstructive pulmonary disease patients. a) Pre-bronchodilator forced expiratory volume in 1 s (FEV1) change on day 14 after treatment with RV568. Data are presented as mean±sem; ANOVA with Bonferonni's multiple comparison to placebo, *: p<0.05. Placebo n=10; 50 μg n=9; 100 μg n=9. b) Sputum supernatant malondialdehyde (MDA) change on day 14 after treatment with RV568. Data are presented as mean±sem; ANOVA with Bonferonni's multiple comparison to placebo, *: p<0.05. Placebo n=6; 50 μg n=7; 100 μg n=4. ICS: inhaled corticosteroids.
There were no changes in sputum cell counts (n=17 patients who produced sputum samples at baseline and day 14). RV568 significantly reduced MDA levels (p<0.05 for both doses); every subject treated with RV568 had reduced MDA levels at day 14 (fig. 5b). There were numerical reductions in CXCL8 and matrix metalloproteinase-1 levels (online supplementary table S7), but no other significant effects of RV568 on sputum biomarkers.
Pharmacokinetics are summarised in online supplementary tables S8 and S9 and further described in the online supplementary material.
Discussion
We have demonstrated anti-inflammatory effects of RV568 on cell and animal models relevant to COPD. We have also shown that RV568 was well tolerated in a 14-day clinical trial, with preliminary evidence for improvement in lung function and anti-inflammatory effects on sputum biomarkers. The pre-clinical data showed that RV568 has greater anti-inflammatory effects than the p38 inhibitor Birb796, which is likely to be due to the different profile of kinases that RV568 targets. Furthermore, RV568 showed greater anti-inflammatory activity than corticosteroids in macrophage and epithelial cell models, and in the cigarette smoke mouse model. The combination of RV568 with corticosteroids brought about anti-inflammatory effects that suggested a synergistic interaction. These results promote the case for longer clinical studies of RV568. We now discuss the findings from pre-clinical and clinical investigations.
Cellular models
We designed experiments to mimic the effects of bacterial infections on monocytes and macrophages using LPS, and the effects of viral infection using epithelial cells stimulated with the TLR3 agonist poly I:C. RV568 exhibited anti-inflammatory effects in both cell models. The epithelial model was particularly corticosteroid resistant, while Birb796 also had no effect. This suggests that RV568 exerted anti-inflammatory effects in this model at least in part by SRC kinase inhibition. SRC is activated following human rhinovirus infection of bronchial epithelial cells, and both TLR3 and ICAM-1 are receptors for human rhinovirus infection [20, 21].
The combination studies with dexamethasone showed synergistic anti-inflammatory effects on cytokine production in poly I:C-stimulated epithelial cells. Birb796 has anti-inflammatory synergistic effects with dexamethasone in LPS-stimulated COPD macrophages [7], and p38 MAPK inhibition can reverse corticosteroid insensitivity in cellular models and cells from asthma patients [22–24]. P38 MAPK phosphorylates the glucocorticoid receptor (GR) [25]. Mercado and co-workers [13, 18] demonstrated that p38 MAPK-α/β and/or MAPK-γ is involved in defective GR nuclear translocation, and RV568 may enhance GR nuclear translocation through modulation of p38 phosphorylation. Our in vitro data suggest potential for RV568 to suppress excessive inflammatory responses to viral infections in COPD patients.
In the monocyte and macrophage models, RV568 consistently showed greater pharmacological effects than Birb796. Previous reports of chemically different p38 inhibitors in primary lung macrophages have shown effective inhibition (Emax approximately 70%) of TNF-α release, but a lower effect on other inflammatory mediators including CXCL8 [26, 27]. Our experiments were similar regarding CXCL8, as Birb796 had minimal effects on CXCL8 secretion from cells differentiated into macrophages (d-THP-1 cells or d-U937 cells) or COPD sputum macrophages. In contrast, RV568 caused significant CXCL8 inhibition, with Emax values >70% in differentiated macrophages, and >50% inhibition in sputum macrophages. RV568 targets p38 MAPK-α and -γ, and differs from Birb796 in its potency against these isoforms. Furthermore, the activity of RV568 against SRC kinases may also contribute to the observed differences; the SRC family kinase is involved in macrophage activation by LPS and oxidative stress [28].
Corticosteroids and RV568 had similar effects in monocyte models and d-U937 cells, with no consistent differences between these drugs noted. However, in differentiated THP-1 cells and COPD macrophages, corticosteroids had minimal effects on CXCL8 secretion. Multiple studies have shown that CXCL8 secretion from COPD and control lung macrophages is corticosteroid insensitive, with <60% inhibition at high corticosteroid concentrations [29, 30]. Furthermore, there is considerable variation between macrophage-derived cytokines in their sensitivity to corticosteroids, although the mechanisms responsible for these differences between cytokines are unclear. RV568 had greater effects than corticosteroids on macrophage-derived CXCL8 production. CXCL8 is a neutrophil chemokine, with increased levels found in COPD lungs [31, 32]; inhibition of CXCL8 overproduction may be of therapeutic benefit.
Effects in mouse models
RV568 showed a dose–response effect on LPS-induced neutrophilic airway inflammation in mice. Administration at 8 h before the LPS challenge resulted in approximately 60% inhibition, indicating prolonged airway activity after a single dose.
The cigarette smoke mouse model is commonly used to mimic the pathophysiological features of COPD. In agreement with previous publications, we found no effect of corticosteroid treatment on the inflammatory response [33, 34]. In contrast, RV568 caused significant inhibition of both neutrophil and macrophage numbers and a range of cytokines. These results demonstrate an ability of RV568 to address cigarette smoke-induced corticosteroid-resistant inflammation. Smoke activates SRC and its inhibition prevents the smoke-induced activation of p38 in epithelial cells [35].
Interestingly, the administration of RV568 with corticosteroid resulted in greater inhibition of inflammation than RV568 alone, suggestive of a synergistic effect and consistent with our cellular results. This in vivo observation suggests the possibility that these drugs, administered concurrently in clinical practice, may enhance overall efficacy.
Clinical trial in COPD patients
The clinical trial was conducted primarily to investigate safety, as RV568 had not been administered to COPD patients previously. We therefore used a small sample size, to limit patient exposure at this early stage of clinical development. There were no safety concerns in this 2-week study.
This study provided an opportunity to explore clinical efficacy, albeit using a small sample size. There was an improvement in FEV1 of 48 mL and 69 mL compared to placebo at 100 μg and 50 μg respectively. The lower dose appeared to cause a greater effect. However, for residual volume an effect of RV568 was only observed at the higher dose. MDA is an oxidative stress biomarker that is elevated in COPD patients [36, 37]. A remarkably consistent MDA reduction was observed in every patient treated with RV568, strongly suggesting an effect on oxidative stress levels. The small patient numbers mean that accurate dose–response information was unlikely to be obtained from these pulmonary function or biomarker measurements. Furthermore, it was not possible to determine which subgroup of patients may derive more benefit, e.g. those taking ICS, or current or ex-smokers. Nevertheless, these results suggest a clinical benefit that should be further investigated in clinical trials of longer duration with larger patient numbers. Given the small numbers of subjects, and short trial duration, we decided not to measure symptom scores using patient-reported outcome tools, as these require larger sample sizes to robustly detect treatment effects.
RV568 had no effect on sputum cells counts, while there was an effect on sputum MDA levels. The inconsistency of the sputum data regarding treatment effects is likely to be due to sample size, as sputum neutrophil measurements in COPD patients are prone to between-day variability [38]. Apart from MDA levels, we did not prove an anti-inflammatory effect of the drug, but the lung function data indicate some pharmacological effects. Overall, we regard these results as preliminary findings of an anti-inflammatory effect in COPD patients requiring validation in larger studies.
Conclusions
Results from cell and animal models relevant to inflammation in COPD showed that RV568 has anti-inflammatory activity that often exceeds the effects of corticosteroids. Furthermore, there was evidence of a synergistic interaction between RV568 and corticosteroids. A small clinical trial provided preliminary evidence of anti-inflammatory and lung function benefits in COPD patients, which requires further investigation in larger, longer term trials.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Disclosures
Acknowledgements
Author contributions: CEC, PR, KI, YK, SL and DS conceived and designed the research; CEC and SL performed the experiments; CEC, KI, CB and SL analysed the data; CEC, PR, KI, YK, CB, SL and DS interpreted the results; CEC, PR and SL prepared the figures; CEC, PR and DS drafted and revised the manuscript; all authors approved the final version.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This report comprises independent clinical research carried out and supported by the National Institute for Health Research (NIHR) South Manchester Respiratory and Allergy Clinical Research Facility at University Hospital of South Manchester NHS Foundation Trust. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received January 26, 2017.
- Accepted July 27, 2017.
- Copyright ©ERS 2017
References










