





Abstract
In this study we investigated the implication of NLRP3 inflammasomes in the pathogenesis of idiopathic pulmonary fibrosis (IPF) and rheumatoid arthritis–usual interstitial pneumonia (RA-UIP).
NLRP3 inflammasome activation at baseline and following stimulation with lipopolysaccharide/ATP was evaluated by measuring interleukin (IL)-1β and IL-18 levels released in the bronchoalveolar lavage fluid (BALF) fluid and by cultures of BALF cells. IL-1β and IL-18 levels were significantly elevated in the BALF and BALF macrophage cultures from RA-UIP patients, consistent with pre-existing inflammasome activation in these patients. In contrast, in IPF, BALF levels of IL-1β were significantly less elevated relative to RA-UIP and IL-18 was lower than controls. Furthermore, upon inflammasome stimulation, IPF BALF macrophage cultures failed to upregulate IL-1β and partly IL-18 secretion, in contrast to controls, which showed robust IL-1β and IL-18 upregulation. Interestingly, RA-UIP BALF cell cultures treated with lipopolysaccharide/ATP showed a potent stimulation of IL-18 secretion but not IL-1β, the latter being already elevated in the unstimulated cultures, while examination of the intracellular IL-1β levels in RA-UIP BALF cells upon NLRP3 inflammasome stimulation showed a significant upregulation of IL-1β suggesting the NLRP3 pathway could be further activated.
Taken together, our results suggest distinct inflammasome activation profiles between autoimmune and idiopathic lung fibrosis.
Abstract
Distinct NLRP3 inflammasome activation profiles between autoimmune and IPF lung macrophages compared with controls http://ow.ly/UKYlp
Introduction
The primary pathogenetic event of idiopathic pulmonary fibrosis (IPF) remains uncertain, despite increased clinical and scientific research in recent years [1, 2]. The median survival from the time of diagnosis is 3 years and explains why IPF is currently considered more lethal than many cancers [3, 4]. The radiologic and histopathologic pattern of IPF is that of usual interstitial pneumonia (UIP) [5]. UIP, unlike the other collagen tissue disorders, is also the most frequent pattern when interstitial lung involvement occurs in patients with rheumatoid arthritis (RA-UIP) [5]. RA-UIP and IPF present some rather intriguing similarities at the clinical and molecular level [5, 6]. Therefore, it is critical to identify initiating events that may drive the pathogenesis of lung fibrosis, both IPF and autoimmune lung fibrosis, with the histologic or radiologic appearance of UIP especially now that we have officially entered a new era with two new agents able to slow IPF progression [7, 8].
Inflammasomes are multimolecular protein complexes responsible for caspase-1-driven activation of the pro-inflammatory cytokine interleukin (IL)-1β [9–11]. They are involved in the innate immunity by recognising pathogen-associated molecular patterns (PAMPs; on bacteria, viruses and fungi), and intracellular and extracellular damage-associated molecular patterns [9]. Furthermore, innate immunity mechanisms have been proposed to have a fundamental role in the initiation of IPF, mainly through the activation of Toll-like receptor (TLR)-9, which drives the rapid progression of fibrotic lung disease through the differentiation of pulmonary fibroblasts into myofibroblasts [12].
Our group has previously shown that primary cell cultures of macrophages from IPF patients infected by herpes simplex virus display a deregulated innate immunity axis in association with increased expression of angiogenesis and fibrogenesis markers [13]. NLRP3 activation could also be impaired in IPF, as is the innate immune response to bacteria and virus infections in IPF fibroblasts and macrophages [3, 4, 13, 14].
Previous studies in animal models have shown that stimulation with asbestos, bleomycin, silica and statins activates inflammasomes and ultimately the production of active IL-1β, which stimulates the production of transforming growth factor (TGF)-β, a central mediator of the development of lung fibrosis, via fibroblast proliferation, collagen production and inhibition of fibroblast apoptosis [15, 16]. Interestingly, it has been postulated that pharmacologically induced NLRP3 activation leads to interstitial lung disease [17]. Caspase-1 activity has also been directly linked to systemic sclerosis-related fibrosis, which suggests that autocrine signalling mediated by IL-1β and IL-18 promotes the pro-fibrotic phenotype observed in these patients [18]. Moreover, NLRP3 inflammasome activation has been linked to neutrophilic asthma [19], while inflammasome-regulated cytokines are critical mediators of acute lung injury [20]. Alternatively, a recent report suggests that the NLRP3 inflammasome pathway is not linked to the severity of stable chronic obstructive pulmonary disease following analyses in the bronchoalveolar lavage fluid (BALF) and cellular components [21]. Recent evidence showing a possible activation of NLRP3 by cigarette smoking, another environmental risk factor for both IPF and rheumatoid arthritis, further supports the possible implication of inflammasomes in lung fibrosis [10, 18]. The role of IL-1β has emerged in the pathogenesis of cigarette smoke-induced emphysema and small airway remodelling [22, 23].
In the current study, we sought to investigate whether NLRP3 inflammasomes are involved in the pathogenesis of lung fibrosis in idiopathic (IPF) and autoimmune (RA-UIP) lung diseases. We used IL-1β and IL-18 cytokine expression in BALF and BALF macrophage cultures as surrogate markers of NLRP3 activation. Our findings suggest distinct inflammasome activation profiles between autoimmune lung fibrosis and IPF.
Patients and methods
Patients
33 consecutive subjects were enrolled in this study in three groups: IPF group (n=16), RA-UIP group (n=8) and control group (n=9). Informed consent was obtained from all patients. The study was approved by the Ethics Committee of the University Hospital of Heraklion, Crete, Greece (17517/19-12-2013).
All patients underwent complete pulmonary function testing (see online supplementary material). No patients had received any immunosuppressive therapy prior to the bronchoscopy.
Biological samples and processing
BALF was obtained from all patients and controls using standard methodology (see online supplementary material). The BALF fractions were pooled and split equally: one sample was sent to the clinical microbiology and cytology laboratory, and the other sample was used for this research, after isolation of the macrophage population, as previously described (see online supplementary material) [13]. BALF cell count was performed using an improved Neubauer chamber.
Equal amounts of BALF sample cells were loaded onto six-well plates using RPMI supplemented with 10% heat-inactivated fetal bovine serum as culture medium.
BALF samples were analysed using standard methodology (see online supplementary material).
Inflammasome activation
TLR-4 stimulation with lipopolysaccharide (LPS, Escherichia coli O111; Merck) (250 pg·mL−1, incubated for 2 h at a 37°C/5% CO2 humidified incubator) was followed by NLRP3 inflammasome activation with a 5 mM ATP pulse (ATP disodium salt hydrate; Sigma-Aldrich) for 20 min at 37°C/5% CO2, in the presence or absence of caspase-1 inhibitor (Calbiochem) (online supplementary figure S1), as previously described [24]. Cells were harvested by centrifugation at 1500×g at 4°C. Supernatants were collected and cells were lysed in RIPA buffer (Sigma-Aldrich), according to the manufacturer's instructions. Supernatants and cell extracts were stored at −80°C.
Total RNA extraction and real-time PCR were performed using standard methodology (see online supplementary material).
Secreted IL-1β, IL-18, TNF-α and IL-6 in all supernatants were assessed by ELISA using commercial kits (eBioscience), according to the manufacturer's instructions. Results were normalised to represent pg·mL−1 per 106 cells.
Intracellular IL-1β protein levels (normalised to actin) were assessed by immunoblotting, as previously described (see online supplementary material).
Statistical analysis
ELISA, immunoblotting as well as quantitative real-time PCR relative expression results were evaluated using the one-sample Kolmogorov–Smirnov goodness-of-fit test and the t-test (unpaired between groups and paired between the conditions in each group). Statistical analysis was carried out using SPSS 17.0 (SPSS, Chicago, IL, USA). Statistical significance was set at the 95% level (p<0.05).
Results
Patient characteristics
Demographics and pulmonary function tests of patients and controls are shown in table 1. Clinical characteristics and pulmonary function tests of the IPF versus RA-UIP group showed no statistical significance, except for sex (mainly females in the RA-UIP group, p=0.003) and forced vital capacity, which was higher in the RA-UIP group (p=0.03). Patients showed no difference in age. Moreover, diffusion capacity was significantly lower in IPF and RA-UIP patients compared with controls (table 1). BALF cell populations showed no difference between IPF and RA-UIP patients (table 2).
Clinical characteristics of the studied groups of patients
Plasma antibodies/antigens and bronchoalveolar lavage fluid (BALF) cell population characteristics of the studied groups of patients
NLRP3 inflammasome-related proteins and pro-inflammatory cytokines in IPF and RA-UIP BALF
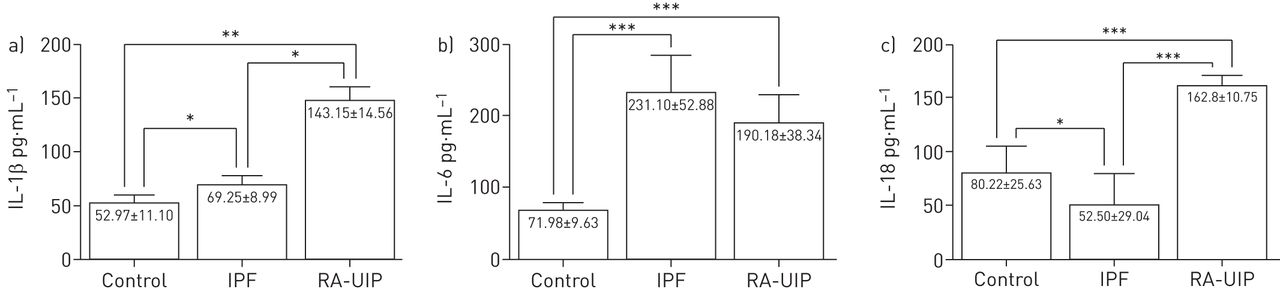
Initially, we measured IL-1β, IL-6 and IL-18 levels in the BALF of IPF and RA-UIP patients in comparison with controls, as an indication of NLRP3 inflammasome activation. We observed significantly elevated levels of IL-1β and IL-18 secreted in the alveolar space of the lungs of RA-UIP patients compared with controls and IPF patients (figure 1a and c), clearly indicating the activation of the inflammasome pathway in RA-UIP. Although not as pronounced as the differences between RA-UIP and controls, IPF BALF showed higher levels of IL-1β and lower levels of IL-18 compared with controls, and therefore was not indicative of inflammasome activation. Interestingly, we observed elevated IL-6 levels in BALF of both IPF and RA-UIP patients, suggesting that other inflammatory pathways in IPF may be active (figure 1b).

a) Interleukin (IL)-1β, b) IL-6 and c) IL-18 levels in the bronchoalveolar lavage fluid of control (n=9), idiopathic pulmonary fibrosis (IPF) (n=16) and rheumatoid arthritis–usual interstitial pneumonia (RA-UIP) (n=8) group patients. Data are presented as mean±sd. *: p<0.05; **: p<0.005; ***: p<0.0005.
Next, we investigated the role of innate immune cells, such as macrophages lining the alveolar epithelium, in the secretion of IL-1β and IL-18 in IPF and RA-UIP patients. The expression of NLRP3 inflammasome components as well as IL-1β and IL-18 production was assessed in unstimulated BALF macrophage cultures (figure 2). We observed that baseline levels of mRNA expression of the NLRP3 inflammasome components NLRP3 and caspase-1 were elevated in both IPF and RA-UIP patients compared with controls (figure 2a and b). Interestingly, however, we observed that, similar to BALF, cultures of BALF macrophages of RA-UIP patients secreted significantly higher amounts of IL-1β as compared with IPF patients and controls (figure 2c). IL-18, however, showed higher values in both patient groups, with IPF having the highest secretion levels (figure 2e). Moreover, IL-6, was elevated in both RA-UIP and IPF patients (figure 2d).

Expression of NLRP3 components and pro-inflammatory cytokines by bronchoalveolar lavage fluid (BALF) macrophages at baseline. Transcript levels of inflammasome components a) NLRP3 and b) caspase-1 in untreated BALF macrophages in control (n=9), idiopathic pulmonary fibrosis (IPF) (n=16) and rheumatoid arthritis–usual interstitial pneumonia (RA-UIP) (n=8) group patients. Concentration of secreted c) interleukin (IL)-1β, d) IL-6 and e) IL-18 in the supernatants of nonstimulated BALF macrophage cultures (baseline) in control (n=9), IPF (n=16) and RA-UIP (n=8) group patients. Data are presented as mean±sd. *: p<0.05; ***: p<0.0005.
Inflammasome activation is impaired in IPF alveolar macrophages
Alveolar macrophages require two signals in order to activate the inflammasome-dependent caspase-1, which results in the release of mature IL-1β and Il-18, a PAMP signal, such as a TLR agonist, and a tissue damage second signal, such as ATP [25]. We performed ex vivo treatments with LPS and ATP in cultures of macrophages from BALF to determine the induction of the NLRP3 pathway in these cells. In addition, since pro-IL-1β cleavage is mediated largely by caspase-1, resulting in the production of IL-1β, we performed NLRP3 activation in the presence of a caspase-1 inhibitor (IL-1β converting enzyme inhibitor I).
In control samples, LPS treatment resulted in a significant increase in IL-1β and IL-18 secretion by 2- and 3.7-fold, respectively (figure 3a and b). However, addition of ATP to the LPS pre-treated cells resulted in a further, significant increase in IL-1β and IL-18 secretion relative to LPS treatment alone by 3.7- and 4-fold, respectively. As expected, IL-1β and IL-18 levels were significantly reduced in the presence of the caspase-1 inhibitor, suggesting a specific activation of the NLRP3 inflammasome in the primary cultures of alveolar macrophages (figure 3a and b). In contrast to IL-1β and IL-18, LPS plus ATP treatment did not result in a significant increase of IL-6 and TNF-α levels relative to LPS treatment alone, as expected (figure 4).
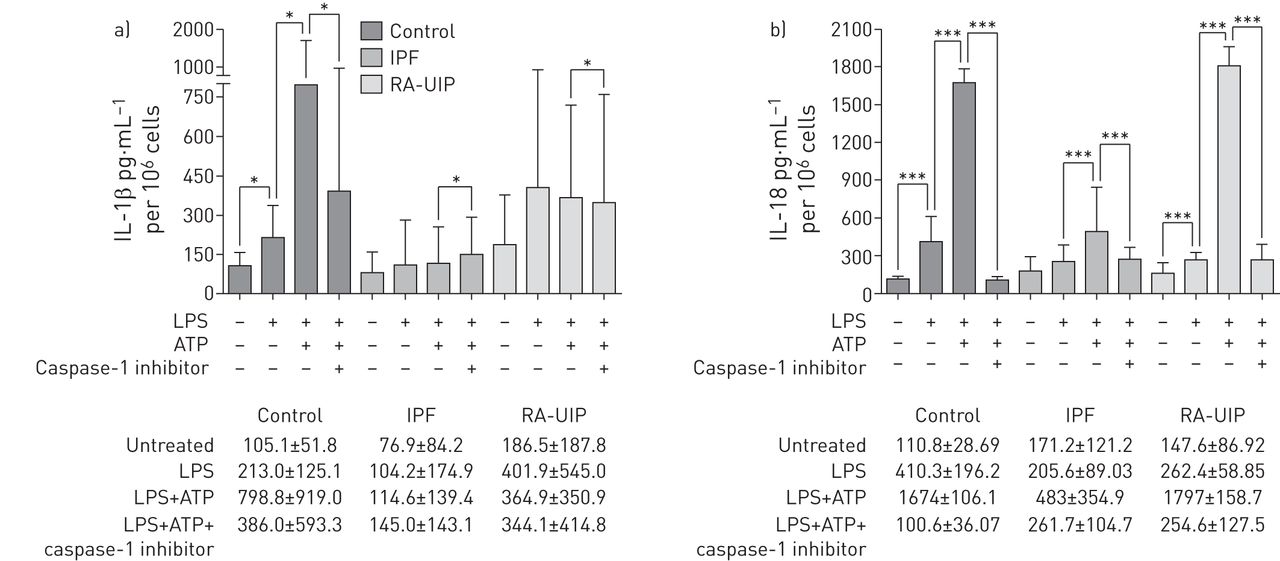
Inflammasome activation in bronchoalveolar lavage fluid (BALF) macrophages of control (n=9), idiopathic pulmonary fibrosis (IPF) (n=16) and rheumatoid arthritis–usual interstitial pneumonia (RA-UIP) (n=8) group patients. a) Concentration of secreted interleukin (IL)-1β in the supernatants of nonstimulated, lipopolysaccharide (LPS)-, LPS+ATP- and LPS+ATP+caspase-1 inhibitor-treated BALF macrophages. b) Concentration of secreted IL-18 in the supernatants of nonstimulated, LPS-, LPS+ATP- and LPS+ATP+caspase-1 inhibitor-treated BALF macrophages. ELISA analysis. Data are presented as mean±sd. *: p<0.05; ***: p<0.0005.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inflammasome activation in bronchoalveolar lavage fluid (BALF) macrophages of control (n=9), idiopathic pulmonary fibrosis (IPF) (n=16) and rheumatoid arthritis–usual interstitial pneumonia (RA-UIP) (n=8) group patients profiles. a) Concentration of secreted interleukin (IL)-6 in the supernatants of nonstimulated, lipopolysaccharide (LPS)-, LPS+ATP- and LPS+ATP+caspase-1 inhibitor-treated BALF macrophages. b) Intracellular protein levels of tumour necrosis factor (TNF)-α in nonstimulated, LPS-, LPS+ATP- and LPS+ATP+caspase-1 inhibitor-treated BALF macrophages. Data are presented as mean±sd. *: p<0.05.
We next treated alveolar macrophages from BALF of IPF and RA-UIP patients, as described for controls. We observed that in IPF, IL-1β levels did not increase following either LPS plus ATP treatment or LPS treatment alone (figure 3a), while IL-18 was significantly upregulated when an ATP pulse was added to the LPS treatment (figure 3b), albeit the induction of IL-18 in IPF was much less pronounced than that observed in controls (figure 3b). In RA-UIP macrophage cultures, we observed that LPS increased the secretion of IL-1β; however, the addition of ATP did not increase IL-1β levels further, as previously observed in the control samples (figure 3a). IL-18, however, was significantly upregulated by LPS plus ATP treatment in RA-UIP macrophage cultures similarly to controls (figure 3b). In addition, IL-6 and TNF-α levels did not change following LPS stimulation in IPF and RA-UIP samples, as was observed in controls (figure 4a and b).
We also analysed the intracellular levels of cleaved IL-1β by immunoblotting, observing that control IL-1β intracellular levels were higher compared with IPF and RA-UIP, suggesting that high amounts of IL-1β are retained in the cytoplasm of control BALF macrophages (online supplementary figure S2). Upon inflammasome activation, IL-1β levels increased in controls while the presence of the caspase-1 inhibitor reduced IL-1β levels. This IL-1β pattern was also observed in RA-UIP samples, but not in IPF samples.
Discussion
The current study investigated the activation of the NLRP3 pathway in BALF from patients with IPF and RA-UIP at baseline and following NLRP3 inflammasome activation.
Our findings suggest distinct inflammasome activation profiles between autoimmune lung fibrosis and IPF. Overall, our results suggest that inflammasome activation is severely impaired in IPF BALF macrophages. Furthermore, the inflammasome activation profile is distinct in RA-UIP, since IL-1β and IL-18 levels are already high at baseline, albeit upregulation is still possible, as shown by the further increase in the newly produced intracellular mature IL-1β levels following LPS plus ATP treatment.
Inflammasomes are activated in RA-UIP lung
We observed that IL-1β and IL-18 levels were elevated in RA-UIP BALF. IL-1β and IL-18 are, at least in part, secreted by alveolar macrophages since we observed that the unstimulated macrophage cultures produced significantly higher levels of these cytokines as compared with controls. In addition, mRNA levels of other components of the NLRP3 pathway such as NLRP3 and caspase-1 were also upregulated in RA-UIP-derived BALF cells. RA-UIP patients were mainly nonsmokers and therefore the apparent inflammasome activation could not be attributed to smoking, a known inflammasome inducer. The elevated levels of IL-1β and IL-18 in RA-UIP patients may also be due to inflammasome-independent mechanisms. Notably, neutrophils are the major producers of inflammasome-independent mature IL-1β [26]. In the RA-UIP group, neutrophil counts were as expected elevated in the BALF samples, which may also account for the elevated IL-1β levels in the BALF of these patients.
IL-6 levels were also elevated in BALF and in the supernatants of BALF cell cultures of RA-UIP patients. The acute release of IL-1β into the peripheral blood in response to cellular stimuli upregulates the expression of various pro-inflammatory cytokines, including IL-6 [4, 5], and it could be hypothesised that this also occurs in the bronchoalveolar spaces in RA-UIP patients.
We observed a trend of IL-1β upregulation upon in vitro stimulation of BALF cells from RA-UIP patients with LPS, whereas intracellular IL-1β production was triggered upon subsequent addition of ATP. It is noteworthy that in controls intracellular IL-1β levels were high in unstimulated and accumulated further in stimulated cells, in contrast to cells from the RA-UIP patients. A recent study showed the accumulation of large amounts of unsecreted IL-1β, retained intracellularly in the LPS-stimulated BALF cells [25], in agreement with our observation in controls. This striking difference is intriguing and a possible explanation could be that intracellular IL-1β levels are depleted due to an elevated NLRP3 inflammasome activation profile in RA-UIP patients. However, since we did observe a strong upregulation of intracellular IL-1β expression upon inflammasome activation in RA-UIP alveolar macrophages, which was completely inhibited in the presence of the caspase-1 inhibitor, we can deduce that the inflammasome pathway can be further activated in these patients.
Impaired activation of the NLRP3 inflammasome in IPF
In IPF, in contrast to RA-UIP, we observed only slightly higher levels of IL-1β in the BALF relative to controls, while cultured BALF cells secreted slightly lower IL-1β levels than controls. In both cases, however, IL-1β levels were significantly lower when compared with RA-UIP samples. Intriguingly, in IPF, similarly to RA-UIP samples, baseline NLRP3 and caspase-1 expression in cultured BALF macrophages as well as IL-18 and IL-6 levels were higher than in the controls. Interestingly, it has been suggested that altered IL-6 signalling in IPF primary fibroblast culture may enhance the resistance of these cells to apoptosis and contribute to a pro-fibrotic effect of IL-6 in IPF [27]. In addition, IL-6 serum levels are suggested to be predictive of early disease progression in patients with systemic sclerosis-associated interstitial lung disease [28].
The main finding of this work is that alveolar macrophages from IPF patients exhibited impaired responses to LPS stimulation and LPS/ATP co-stimulation, leading to the hypothesis that both TLR-4 and NLRP3 inflammasome pathways are dysregulated. In particular, we observed no NLRP3-specific induction of either intracellular or secreted IL-1β in alveolar macrophage cultures in IPF, while the induction of IL-18 was significantly lower in IPF relative to RA-UIP and controls.
Our observations are in agreement with cumulative evidence that the innate immune response is impaired in patients with IPF, including the reduced functional capacity of macrophages to kill bacteria [29]. Additionally, recent data from our group and others suggest that viruses are the principal cofactors in the initiation and progression of IPF [13, 30, 31]. Our present findings suggest that deregulation of inflammasome activation in fibrotic patients could be crucial in the progression of the disease, in particular with respect to the occurrence of acute exacerbations.
A possible mechanism for the reduced triggering of inflammasome activity could be impaired autophagy, which interferes with mitochondrial turnover, stimulates NLRP3 activation and predisposes for IL-1β hypersecretion through mitochondria-generated reactive oxygen species [32, 33]. Autophagy and mitophagy appear to be defective in IPF lungs [34–36], suggesting a possible explanation for the exhaustion of the inflammasome machinery in pulmonary fibrosis.
Immune mechanisms have been implicated in IPF [37] and are also central to the development of rheumatoid lung. However, autoimmunity in IPF needs to be interpreted with caution, as it could provide a possible future therapeutic target. Recently, a potential beneficial effect of rituximab in interstitial lung disease associated with connective tissue disease [38] has been highlighted. Our study provides further evidence for the association of innate immunity with IPF; however, this effect is more prominent, as expected, in rheumatoid lung. This finding is in agreement with emerging molecular data linking immune molecules such as CD28 with poor outcome in IPF [39]. As the inflammasome plays a central role in host defence against wide variety of infectious and noninfectious agents, this finding further supports the dysregulation of the innate immune response of IPF patients which has been linked to the pathogenesis of IPF and in particular the inability of these patients to resolve lung infections linked to acute exacerbations [20].
The importance of inflammation in initiation and progression of pulmonary fibrosis has already been established [40], while the initial pro-inflammatory scenario of inflammasome activation can rapidly progress to a fibrotic one. Our findings reveal a significant lapse in NLRP3 inflammasome activation in alveolar macrophages in IPF, while suggesting an impaired activation of NLRP3 in autoimmune fibrosis, compared with healthy macrophages. We hypothesise that lung external stimuli activating the NLRP3 inflammasome exert less influence on IPF macrophages than RA-UIP macrophages, although further research is essential in establishing an etiopathological link between NLRP3 inflammasome processes and IPF. It is thus imperative to explore all possible pathological and therapeutic implications of inflammasomes in the fibrotic lung.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This project has been supported by a Hellenic Thoracic Society grant. Funding information for this article has been deposited with FundRef.
Conflict of interest: None declared.
- Received April 9, 2015.
- Accepted November 9, 2015.
- Copyright ©ERS 2016
References










