



Inhibition of Pim1 kinase, new therapeutic approach in virus-induced asthma exacerbations
- Maaike de Vries1,2,3⇑,
- Nicole Bedke2,
- Natalie P. Smithers2,
- Matthew Loxham2,
- Peter H. Howarth2,4,
- Martijn C. Nawijn1,3 and
- Donna E. Davies2,4
- 1University of Groningen, University Medical Center Groningen, Experimental Pulmonology and Inflammation Research, Dept of Pathology and Medical Biology, Groningen, The Netherlands
- 2Brooke Laboratory, Academic Unit of Clinical and Experimental Sciences, University of Southampton, Faculty of Medicine, University Hospital Southampton, Southampton, UK
- 3University of Groningen, University Medical Center Groningen, GRIAC Research Institute, Groningen, The Netherlands
- 4National Institute for Health Research, Respiratory Biomedical Research Unit, University Hospital Southampton, Southampton, UK
- Maaike de Vries, Laboratory of Experimental Pulmonology and Inflammation Research, Department of Pathology and Medical Biology, University Medical Center Groningen, Hanzeplein 1, 9713 GZ, The Netherlands. E-mail: m.de.vries04{at}umcg.nl
Abstract
Therapeutic options to treat virus-induced asthma exacerbations are limited and urgently needed. Therefore, we tested Pim1 kinase as potential therapeutic target in human rhinovirus (HRV) infections. We hypothesised that inhibition of Pim1 kinase reduces HRV replication by augmenting the interferon-induced anti-viral response due to increased activity of the janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway.
Air–liquid interface (ALI) cultures of primary bronchial epithelial cells (PBECs) from healthy individuals and moderate-to-severe asthmatic volunteers were infected with HRV-16 with or without a specific Pim1 inhibitor; viral replication and induction of anti-viral responses were measured using RT-qPCR. Viral titres were measured by 50% tissue culture infective dose and release of interferon-γ-induced protein 10 (IP-10) and RANTES protein assessed by ELISA. Phosphorylation of STAT-1 was determined using western blotting.
Viral replication was reduced in ALI cultures of healthy and asthmatic PBECs treated with the Pim1 inhibitor. Using cultures from healthy donors, enhanced STAT-1 phosphorylation upon inhibition of Pim1 kinase activity resulted in increased mRNA expression of interferon-β, interleukin-29, IP-10 and RANTES 12 h after infection and increased protein levels of IP-10 and RANTES 24 h after infection.
We have identified Pim1 kinase as novel target to reduce viral replication in ALI cultures of PBECs. This may open new avenues for therapeutic interventions in virus-induced asthma exacerbations.
Abstract
Pim1 inhibition reduces HRV-16 replication, implying a new therapeutic approach for virus-induced asthma exacerbations http://ow.ly/Xxmy2
Introduction
Asthma is a chronic respiratory disease, affecting up to 10% of the population in the Western world [1]. It is characterised by reversible airflow obstruction, airway inflammation and airway hyperresponsiveness, and is further complicated by frequent occurrence of exacerbations [1, 2]. The main triggers of asthma exacerbations are infections with respiratory viruses, most notably human rhinoviruses (HRVs); and it has been suggested that patients with asthma are more susceptible towards HRV infections compared with non-asthmatic individuals [3]. Asthma exacerbations are a major economic and social burden and therapeutic options for the treatment of virus-induced asthma exacerbations are limited to date, emphasising the pressing need for novel therapeutics [1].
In the lower airways, the principal target cells for infection with HRV are bronchial epithelial cells. These cells form the first line of defence against potential harmful pathogens by constituting a physical and immunological barrier covering the entire surface of the respiratory tract [1, 4, 5]. HRV infection of epithelial cells induces the expression of type I and III interferons (IFNs) IFN-β and interleukin (IL)-29, respectively, which in turn can activate the janus kinase–signal transducer and activator of transcription (JAK-STAT) signalling pathway [4–8]. Subsequent transcription of a wide range of interferon-inducible genes results in reduced viral replication and limitation of viral spread [6]. It has been shown that patients with asthma display an impaired interferon response upon infection with HRV resulting in increased viral replication compared with healthy individuals [9, 10]. Notwithstanding, primary bronchial epithelial cells (PBECs) of asthmatic volunteers exhibit a normal cellular response to IFN-β, and exogenous administration of IFN-β has been postulated as a viable therapeutic treatment in virally induced asthma exacerbations [11]. Indeed, recent clinical trials have shown beneficial effects of inhaled IFN-β in viral-induced exacerbations in patients with difficult-to-treat asthma [12]. These findings support the concept of identifying approaches that augment the induction of an interferon-induced anti-viral response as novel therapeutic strategy.
The interferon-activated JAK-STAT signalling pathway is negatively regulated by suppressor of cytokine signalling (SOCS) proteins [13, 14]. Expression of SOCS1 and SOCS3 was found to be increased by HRV infection of PBECs in a transforming growth factor (TGF)-β-dependent fashion [15], suggesting a contribution of SOCS proteins to desensitisation of HRV infected PBECs to type I interferon responses. SOCS proteins can be stabilised by phosphorylation through the Pim family of serine/threonine kinases, resulting in reduced proteosomal degradation of SOCS proteins, which subsequently leads to further reduction of JAK-STAT signalling [16]. Pim kinases are involved in a wide range of cellular processes like cell differentiation, cell survival and cytokine signalling and we have previously shown that Pim1 kinase is highly expressed in bronchial epithelial cells [17, 18]. Therefore, we hypothesised that inhibition of Pim1 kinase reduces HRV replication in PBECs by augmenting the interferon-induced anti-viral response due to increased activity of the JAK-STAT signalling pathway.
In this study, we test our hypothesis by evaluating the effect of Pim1 kinase inhibition on viral replication in air–liquid interface (ALI) cultures of PBECs derived from healthy individuals and moderate-to-severe asthmatic volunteers and assess the potential of pharmacological inhibition of Pim1 kinase activity as new therapeutic approach in virally induced asthma exacerbations.
Methods
A full description of the methods can be found in the online supplement.
Cell cultures
Bronchial brushings were obtained from subjects without or with asthma by fibre-optic bronchoscopy according to standard guidelines. All procedures were ethically approved by the Southampton and South West Hampshire Research Ethics Committees (REC no. 05/Q1702/165 and REC no. 09/H0504/109). Written informed consent was received from all volunteers. Detailed subject characteristics can be found in table S1 in the online supplement.
Brushings were processed for PBEC culture in bronchial epithelium growth medium, as previously described [19, 20]. When sub-confluent, cells were seeded on transwells and cultured on ALI for 21 days. Fully differentiated ALI cultures were starved in basal medium and pre-treated with 5 µM Pim1 inhibitor K00135 when indicated [21, 22].
Human rhinovirus-16
Human rhinovirus-16 (ATCC VR-283™, Teddington, UK) was amplified in H1HeLa cells as described previously [23]. The viral titre was determined by 50% tissue culture infective dose (TCID50) at 1.76×107 per mL and multiplicity of infection (MOI) of 5 was used for all infections.
Viral infection
ALI cultures of PBECs were apically infected for 6 h at 33°C, where after the cultures were washed apically twice with HBSS and the apical and basolateral medium, containing Pim1 inhibitor when indicated, was replaced. As controls, medium, UV-irradiated HRV-16 (1200 mJ·cm−2 for 50 min on ice) and Pim1 inhibitor alone were included.
RNA isolation, reverse transcription and RT-qPCR
ALI cultures were harvested in TRIzol reagent (Invitrogen, Paisley, UK) and total RNA isolated following standard protocols. Contaminating DNA was enzymatically removed (DNA-free kit, Ambion, Austin, TX, USA) and reverse transcription was performed with NanoScript reverse transcription kit (PrimerDesign Ltd, Southampton, UK).
mRNA expression levels were determined by RT-qPCR using 20 ng cDNA template and calculated relative to the average of the housekeeping genes GAPDH and UBC. A cloned synthetic DNA construct packaged at known copy number was used for determination of the viral copy numbers of HRV-16.
Analysis of viral particles and cytokines
The release of viral particles in the supernatant was determined using TCID50 assay [9]. The levels of IFN-γ-induced protein 10 (IP-10) and RANTES in the apical supernatant were determined by ELISA (R&D Systems, Abingdon, UK), according to manufacturer's instructions with half of the recommended volumes used for every step.
Western blotting
ALI cultures were lysed in sample buffer and analysed by SDS-PAGE. Antibodies and their dilutions are summarised in the online supplement.
Statistical analysis
Statistical analysis was performed with Graphpad Version 6.01. The data were not normally distributed and were analysed using the two-tailed non-parametric Wilcoxon matched-pairs signed rank test for the healthy individuals and the one-tailed non-parametric Wilcoxon matched-pairs signed rank test for the asthmatic individuals.
Results
Inhibition of Pim1 kinase activity reduces viral replication in ALI cultures
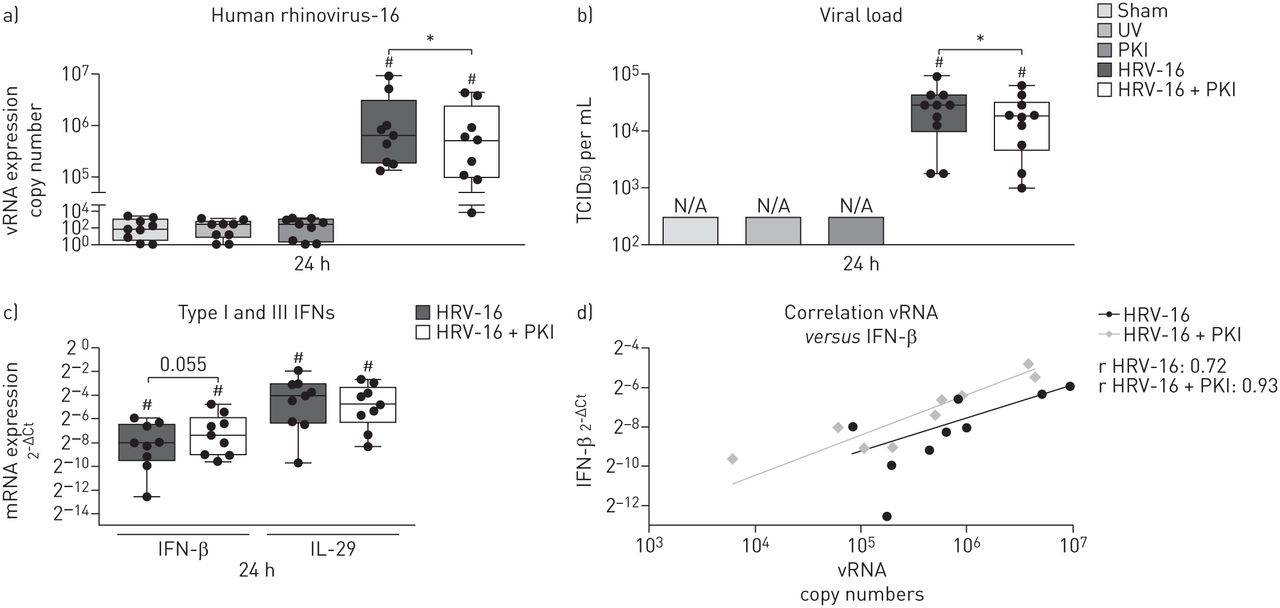
Fully differentiated ALI cultures were infected with HRV-16 and significantly increased viral copy numbers were observed 24 h after infection compared to the appropriate controls (figure 1a). Interestingly, concurrent inhibition of Pim1 kinase resulted in significantly decreased viral copy numbers compared with the cultures infected with HRV-16 alone. Subsequently, we analysed the release of viral particles in the supernatant 24 h after infection and, consistent with the reduced viral copy numbers, inhibition of Pim1 kinase resulted in a decreased release of viral particles into the supernatant compared with the control cultures (figure 1b). Although the differences in response to viral infection were considerable between the individuals, the overall reduction in viral replication and release of viral particles was 20% and 35%, respectively (online supplement figure S1).

Inhibition of Pim1 kinase activity reduces viral replication in air–liquid interface (ALI) cultures. ALI cultures of primary bronchial epithelial cells from healthy volunteers, pre-treated with the Pim1 inhibitor (PKI) or control, were infected with human rhinovirus (HRV)-16 at multiplicity of infection of 5 for 6 h at 33°C and cultured until 24 h after the start of infection. ALI cultures were analysed for a) viral (v)RNA copy numbers, b) viral load (measured by 50% tissue culture infective dose (TCID50)) and c) mRNA expression of interferon (IFN)-β and interleukin (IL)-29. d) Correlation between vRNA copy number and mRNA expression of IFN-β. Median and range of nine donors are shown. Statistical significance was tested with the paired two-sided Wilcoxon matched-pairs signed rank test and p<0.05. *: p<0.05; #: p<0.05 compared with sham-infected control.
Analysis of mRNA expression of the Type I and III interferons 24 h after infections revealed that infection with HRV-16 significantly induced the expression of IFN-β and IL-29 compared to sham-infected control cultures (figure 1c). The Pim1 kinase inhibitor itself did not have an effect on the expression of either IFN-β and IL-29 (data not shown). While inhibition of Pim1 kinase resulted in a trend towards increased mRNA expression levels of IFN-β, no differences in expression of IL-29 upon viral infection in the presence of the Pim1 kinase inhibitor were observed. However, even though interferon levels were not significantly increased, the levels of IFN-β expression were enhanced relative to the viral load within the cell (figure 1d).
Taken together, these data suggest that pharmacological inhibition of Pim1 kinase activity reduces viral replication in ALI cultures of PBECs from healthy individuals in association with maintenance of a high level of IFN expression, even upon reduced viral replication.
Inhibition of Pim1 kinase activity reduces viral replication in ALI cultures of PBECs from asthmatic volunteers
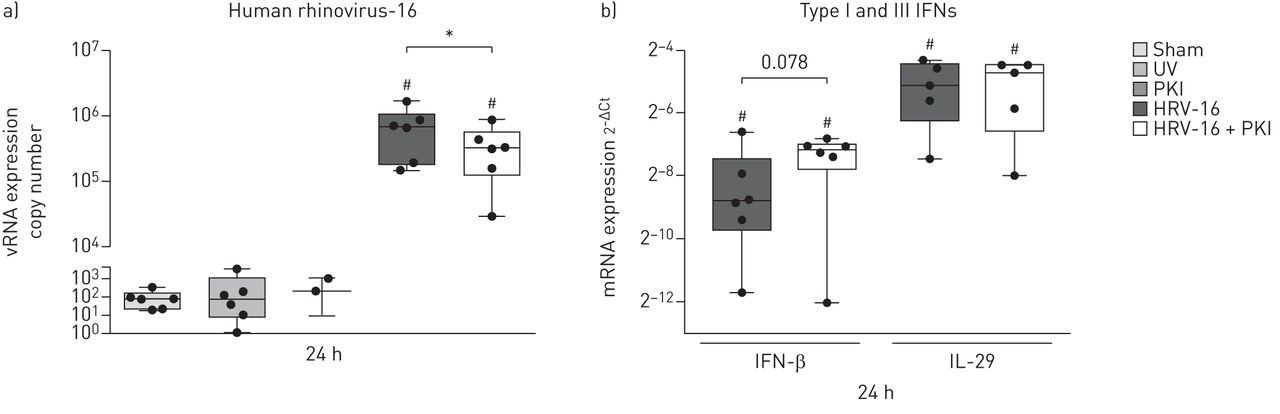
Since virally induced exacerbations are particularly problematic for asthmatic patients, we determined the effects of inhibition of Pim1 kinase activity on viral replication and anti-viral response in ALI cultures of PBECs from moderate-to-severe asthmatic volunteers. In the presence of the Pim1 kinase inhibitor, we observed significantly decreased viral copy numbers compared to the cultures infected in the absence of the Pim1 kinase inhibitor (figure 2a). With the relatively small number of donors studied, no differences in viral replication between healthy individuals and moderate-to-severe asthmatic patients could be observed (online supplement figure S2). In line with our observations in the virally infected ALI cultures of PBECs from healthy individuals, infection with HRV-16 resulted in marked induction of IFN-β and IL-29 expression. As observed in the PBECs from healthy individuals 24 h after infection, concurrent inhibition of Pim1 kinase resulted in a trend (p=0.078) towards increased IFN-β mRNA expression levels, while IL-29 mRNA expression was unaffected upon inhibition of Pim1 kinase activity (figure 2b). This effect was evident in four out of six cases where treatment with the Pim1 inhibitor augmented IFN-β levels (online supplement figure S3a), while viral RNA levels were reduced. In the subject with the lowest and unaffected IFN-β expression, viral copy number was low and did not change in the presence of the inhibitor (online supplement figure S3b). These data suggest that, in most cases, inhibition of Pim1 kinase can also reduce viral replication in ALI cultures of PBECs from moderate-to-severe asthmatic volunteers with maintenance of the anti-viral response upon reduced viral replication.

Inhibition of Pim1 kinase activity reduces viral replication in air–liquid interface (ALI) cultures of primary bronchial epithelial cells (PBECs) from asthmatic volunteers. ALI cultures of PBECs from moderate-to-severe asthmatic volunteers, pre-treated with the Pim1 inhibitor (PKI) or control, were infected with human rhinovirus (HRV)-16 at multiplicity of infection of 5 for 6 h at 33°C and cultured until 24 h after the start of infection. ALI cultures were analysed for a) viral (v)RNA copy numbers and b) mRNA expression of interferon (IFN)-β and interleukin (IL)-29. Median and range of six donors are shown for vRNA copy numbers and expression of IFN-β and for five donors for the expression of IL-29. Statistical significance was tested with the paired one-sided Wilcoxon matched-pairs signed rank test and p<0.05. *: p<0.05; #: p<0.05 compared with sham-infected control.
Inhibition of Pim1 kinase activity induces an early anti-viral response
To elucidate the mechanism behind the reduced viral replication upon inhibition of Pim1 kinase activity, we analysed viral replication and the subsequent activation of the anti-viral response at earlier times after infection in ALI cultures of PBECs from healthy individuals.
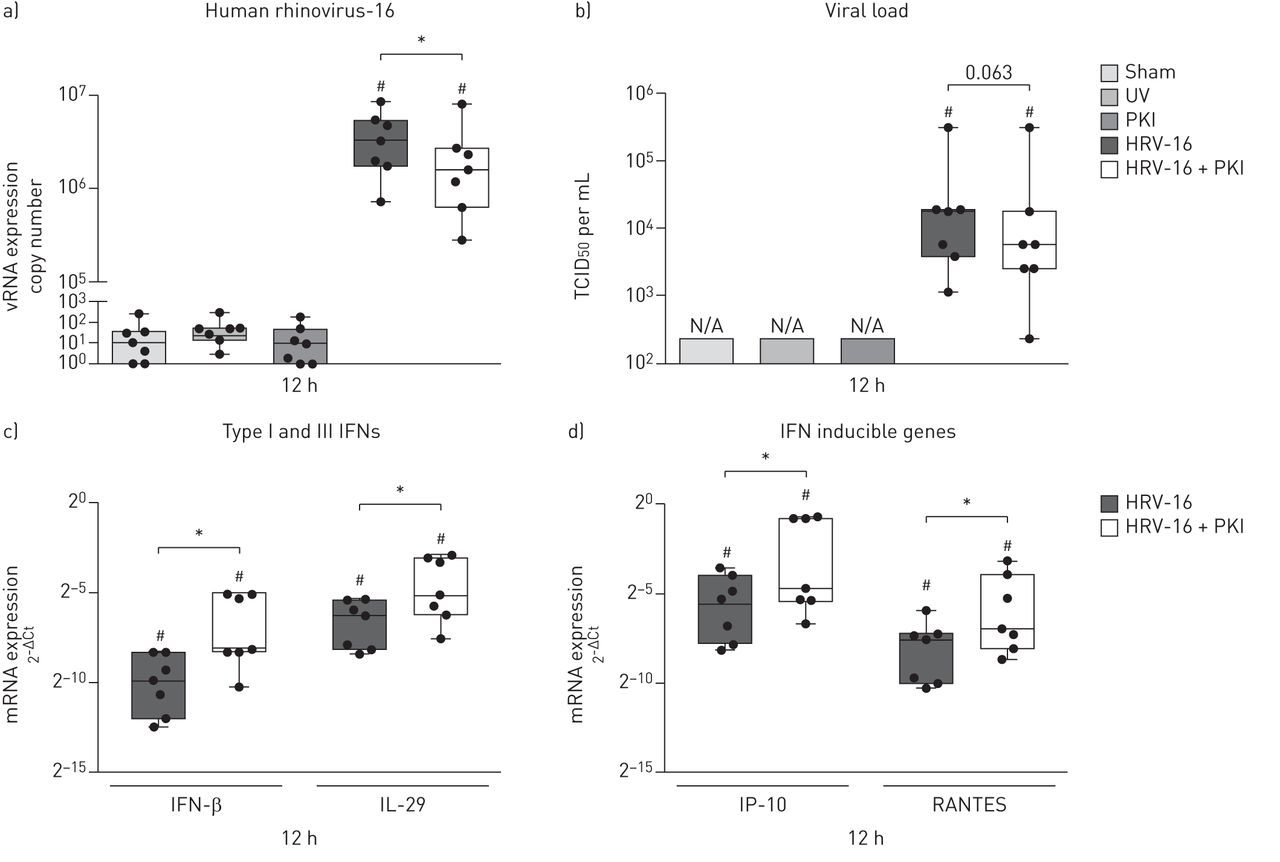
Infection with HRV-16 resulted in highly increased viral copy numbers compared with control-treated ALI cultures 12 h after infection, which was significantly reduced with 50% in the presence of the Pim1 kinase inhibitor (figure 3a and online supplement figure S4a). As shown in figure 3b, by this time, viral particles were detectable in the apical supernatant of the infected ALI cultures. Here, we observed a trend (p=0.063) towards a decrease in the release of viral particles of approximately 70% in the presence of the Pim1 kinase inhibitor (online supplement figure S4b).

Inhibition of Pim1 kinase activity induces an early anti-viral response. Air–liquid interface (ALI) cultures of primary bronchial epithelial cells from healthy volunteers, pre-treated with the Pim1 inhibitor (PKI) or control, were infected with human rhinovirus (HRV)-16 at multiplicity of infection of 5 for 6 h at 33°C and cultured until 12 h after the start of infection. The cultures were analysed for a) viral (v)RNA copy numbers, b) viral load (measured by 50% tissue culture infective dose (TCID50)), c) mRNA expression of interferon (IFN)-β and interleukin (IL)-29 and d) IFN-γ-induced protein 10 (IP-10) and RANTES. Median and range of seven donors are shown. Statistical significance was tested with the paired two-sided Wilcoxon matched-pairs signed rank test and p<0.05. *:p<0.05; #: p<0.05 compared with sham-infected control.
Analysis of the expression of IFN-β and IL-29 showed strongly enhanced mRNA levels upon viral infection compared to mock-infected controls. Concurrent inhibition of Pim1 kinase activity further augmented the mRNA expression of both IFN-β and IL-29 (figure 3c), while inhibition of Pim1 kinase in the absence of viral infection did not have an effect on the expression of IFN-β and IL-29 (data not shown). Subsequent analysis of the mRNA expression of the interferon inducible genes IP-10 and RANTES showed a comparable effect: infection with HRV-16 induced the expression of IP-10 and RANTES, which was significantly enhanced in the presence of the Pim1 kinase inhibitor (figure 3d), but not by the inhibitor alone.
To summarise, these data demonstrate that inhibition of Pim1 kinase augments the early anti-viral response, resulting in enhanced expression of IFN-β and IL-29 as well as their target genes IP-10 and RANTES, thereby leading to reduced viral replication.
Increased apical release of IP-10 and RANTES upon inhibition of Pim1 kinase activity
To establish the mRNA expression data of IP-10 and RANTES seen 12 h after infection, we measured the protein levels of these cytokines in the supernatant of the infected cultures. After 24 h, inhibition of Pim1 kinase activity resulted in significantly increased levels of apically released IP-10 (figure 4a) and RANTES (figure 4b) compared with HRV-16 infection alone. The levels of IP-10 and RANTES were increased after 48 h compared with the levels detected 24 h after infection, but no differences could be observed upon inhibition of Pim1 kinase activity, reflecting the similar levels of IFN expression observed at 24 h post infection (figure 1c). These data confirm that inhibition of Pim1 kinase activity results in enhanced anti-viral activity as evidenced by increased apical release of IP-10 and RANTES 24 h after infection.
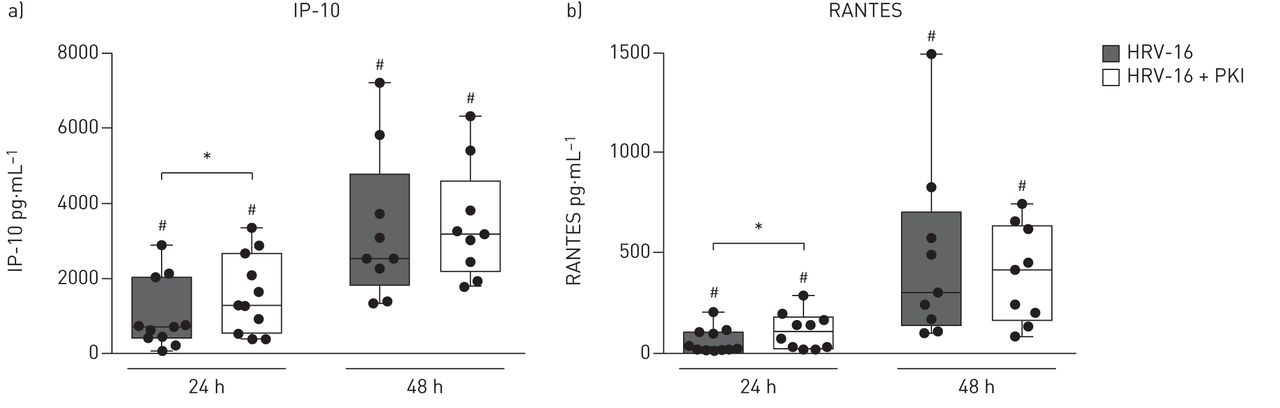
Increased apical release of interferon (IFN)-γ-induced protein 10 (IP-10) and RANTES upon inhibition of Pim1 kinase activity. Air–liquid interface (ALI) cultures of primary bronchial epithelial cells for healthy volunteers, pre-treated with the Pim1 inhibitor (PKI) or control, were infected with human rhinovirus (HRV)-16 at multiplicity of infection of 5 for 6 h at 33 °C and cultured until 24 and 48 h after the start of infection. The amount of apically released a) IP-10 and b) RANTES was determined with ELISA, 24 and 48 h after infection. Median and range of 9–11 donors are shown. Statistical significance was tested with the paired two-sided Wilcoxon matched-pairs signed rank test and p<0.05. *:p<0.05; #: p<0.05 compared with sham-infected control.
Inhibition of Pim1 kinase activity induces phosphorylation of STAT-1
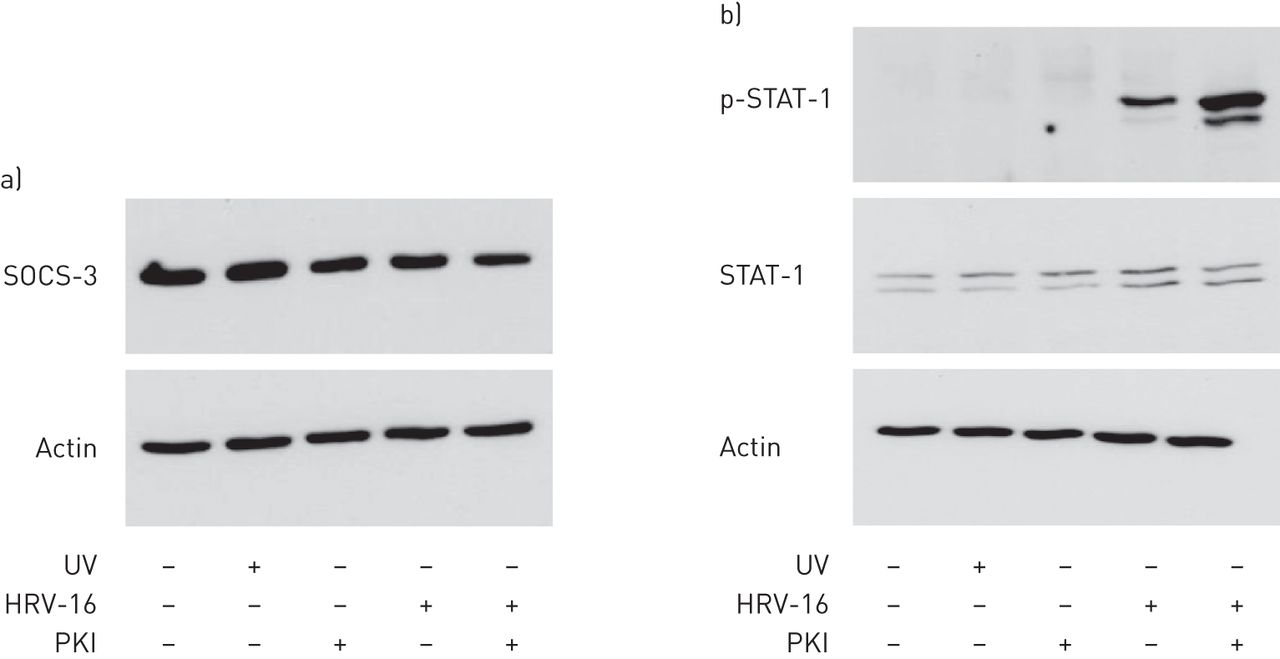
Since we previously showed that inhibition of Pim1 kinase reduced viral replication by enhancing the induction of cell death in monolayer cultures of PBECs [22], we first assessed the role of cell death in ALI cultures. We could not detect leakage of lactate dehydrogenase (LDH) into the apical or basolateral compartments of the ALI cultures and therefore, we analysed the protein levels of cleaved cytokeratin-18 and cleaved poly-(ADP-ribose) polymerase as markers for apoptosis. We revealed a minimal evidence for the induction of apoptotic cell death in the epithelial cell cultures 12 and 24 h after infection, irrespective of treatment conditions (online supplement figure S5). Therefore, we continued by evaluating the effect of Pim1 kinase inhibition on the anti-viral response. To test if augmented proteosomal degradation of SOCS proteins upon inhibition of Pim1 kinase as postulated by Chen et al. [16] was causative in the enhanced interferon signalling, we sought to measure the levels of SOCS-3 in the cell. Unfortunately, we were not able to detect significant differences in the levels of SOCS-3 proteins in cell lysates of ALI cultures 12 h after infection (figure 5a). In addition, SOCS-1 was only detectable at mRNA level and since the expression of SOCS-1 seems to be interferon-dependent, the effect of inhibition of Pim1 kinase is not likely to be critically regulated by SOCS-1 (online supplement figure S6). Therefore, we decided to test downstream activation of the interferon-signalling pathway and analysed the phosphorylation of STAT-1. Phosphorylated STAT-1 was only detectable in virally infected ALI cultures, while total STAT-1 was detected at similar levels in all the cultures (figure 5b). Inhibition of Pim1 kinase activity resulted in strongly increased phosphorylation of STAT-1 compared with the HRV-16 infected cultures alone.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Augmented phosphorylation of signal transducer and activator of transcription (STAT)-1 upon inhibition of Pim1 kinase activity. Air–liquid interface (ALI) cultures of primary bronchial epithelial cells from healthy volunteers, pre-treated with the Pim1 inhibitor (PKI) or control, were infected with human rhinovirus (HRV)-16 at multiplicity of infection of 5 for 6 h at 33 °C and cultured until 12 h after the start of infection. Protein levels of a) suppressor of cytokine signalling (SOCS)-3 and b) (phosphorylated) STAT-1 were determined by western blotting. Data are representative of four independent experiments. p: phosphorylated.
Discussion
In this study, we assessed the potential of Pim1 kinase as novel target for therapeutic intervention in virally induced asthma exacerbations, using an HRV-16 infection model in PBECs cultured under ALI conditions. We demonstrated that 24 h after infection, viral replication was reduced in ALI cultures of PBECs from healthy individuals and moderate-to-severe asthmatic volunteers treated with a pharmacological Pim1 kinase inhibitor. The reduced viral replication was accompanied by an augmented anti-viral inflammatory response characterised by increased gene expression of IFN-β, IL-29, IP-10 and RANTES, 12 h after infection and enhanced release of IP-10 and RANTES, 24 h after infection. Western blot analysis of the phosphorylation status of STAT-1 revealed that inhibition of Pim1 kinase activity in virally infected ALI cultures of PBECs led to increased activation of STAT-1, 12 h after infection, reflecting an increased type-I interferon signalling. These data suggest that Pim1 kinase suppresses the innate anti-viral response and that inhibition of its activity may thus offer a new therapeutic approach for virus-induced asthma exacerbations.
Asthmatic subjects suffer from exacerbations of their disease with one of the main triggers being respiratory virus infection [3]. The most commonly detected viruses are human rhinoviruses, of which more than 100 serotypes have been identified, precluding efficient design of vaccination strategies against HRVs [24]. While asthma has not been shown to be a risk factor for the development of respiratory infections upon infection with HRV, the more frequent, more severe and longer lasting symptoms of lower-respiratory-tract infections observed in asthmatic patients compared to healthy individuals suggest an increased susceptibility towards rhinovirus infection [25]. The underlying mechanism for this enhanced susceptibility of asthma patients to rhinovirus infections has been associated with a deficiency of the innate immune response in the bronchial epithelium [9, 10]. In the present study, we infected fully differentiated ALI cultures of PBECs from moderate-to-severe asthmatic patients and healthy individuals with human rhinovirus. Exposing PBECS to an air interface in the presence of retinoic acid for 21 days results in a pseudostratified epithelium containing ciliated, goblet and basal cells, recapitulating the airway surface in vivo [26]. Although it has been shown before that differentiated PBECs are more resistant towards infection with HRV compared to the cells of monolayer cultures [27, 28], we achieved unambiguous viral infection using a multiplicity of infection of 5, as assessed by viral replication and induction of the anti-viral responses in cultures from both healthy and moderate-to-severe asthmatic individuals. While studies by Wark et al. [9] and Contoli et al. [10] using monolayer cultures of PBECs from moderate-to-severe asthmatic subjects revealed increased susceptibility of asthmatic patients towards viral infection, this was not observed in cultures from mild asthmatic subjects [29]. Although we did not observe significant differences in viral replication between differentiated cultures from healthy individuals and asthmatic volunteers, which is in line with previous studies in fully differentiated cultures [30, 31], our studies were not powered to show such a difference.
We recently found that higher endogenous levels of TGF-β in PBEC cultures from asthmatic volunteers augmented viral replication and suppressed IFN expression in association with increased gene expression levels of SOCS-1 and SOCS-3 [15]. SOCS proteins are negative regulators of interferon signalling by suppressing the intensity and duration of JAK-STAT activation [13, 14]. In addition to transcriptional regulation, SOCS proteins can bind to elongin BC E3 ligases, leading to their ubiquitin-mediated proteosomal degradation [32]. Of note, binding to elongin BC E3 ligases can be prevented by phosphorylation of SOCS proteins by Pim kinases, thereby reducing their degradation and enhancing their suppressive activity towards JAK-STAT signalling [16]. Thus, we postulate that inhibition of Pim1 kinase will enhance the binding of SOCS proteins to elongin BC E3 ligases and subsequently increase the proteosomal degradation of SOCS proteins. Overall, this will lead to augmented JAK-STAT activation and increase the interferon-induced anti-viral response. Although we were not able to show decreased total levels of SOCS-3 protein in the presence of the Pim1 kinase inhibitor, this might be explained by relatively high basal levels of SOCS proteins impeding accurate quantification of differences upon viral infection and inhibition of Pim1 kinase activity. Nonetheless, we found augmented phosphorylation of STAT-1 in virally infected ALI cultures treated with the Pim1 inhibitor, suggesting that inhibition of Pim1 kinase activity indeed resulted in enhanced activation of the JAK-STAT pathway. This activation was further established by increased expression levels of IFN-β and IL-29, and increased expression of the interferon inducible genes IP-10 and RANTES at transcriptional and protein levels observed upon inhibition of Pim1 kinase activity in virally infected ALI cultures.
As observed with monolayer cultures pre-treated with IFN-β to augment the anti-viral response [9, 11], we observed reduced replication of HRV-16 in association with an augmented anti-viral response upon inhibition of Pim1 kinase activity using ALI cultures from either healthy individuals or moderate-to-severe asthmatic volunteers. These observations reveal that inhibition of Pim1 kinase activity can reduce viral replication, irrespective of the disease status, suggesting that inhibition of Pim1 kinase might be an interesting new therapeutic approach in the treatment of virus-induced asthma exacerbations.
Notwithstanding the promising results of inhibition of Pim1 kinase activity in reducing viral replication in ALI cultures of healthy individuals and asthmatic volunteers, we have to consider some limitations of our study. One limitation of our study is the use of pharmacological inhibition of Pim1 kinase activity. Although the Pim1 kinase inhibitor K00135 is highly specific and widely used in Pim1 kinase research [21, 33, 34], we cannot exclude the occurrence of off-target effects. However, the complexity of our in vitro model of ALI cultures of PBECs complicates the establishment of pharmacological inhibition of Pim1 kinase activity with for example siRNA constructs to knock-down the activity at transcriptional level. Indeed, even using monolayer cultures of PBECs, we were unable to achieve significant reduction of Pim1 kinase protein, even though good knock down was observed at the mRNA level (online supplement figure 7). Therefore, to confirm that the reduced viral replication in the presence the Pim1 kinase inhibitor was not merely an off-target effect of the inhibitor, we also tested a structurally unrelated pharmacological Pim1 kinase inhibitor, that has been validated in literature for effective inhibition of Pim1 kinase activity both in vitro and in vivo [35]. As shown in online supplement figure 8, viral replication and viral load in PBECs were also inhibited by SMI-4a suggesting that the effects on viral replication are mediated by inhibition of Pim1 kinase. A second limitation of our study is the single use of the major group virus HRV-16 to induce viral infections in bronchial epithelial cells. Further studies should reveal if the replication of other HRV strains, including not only minor group viruses but also HRV-C, could be reduced upon inhibition of Pim kinase activity. Next to HRV, influenza and respiratory syncytial viruses are commonly detected in exacerbations and it would be worthwhile to establish if inhibition of Pim1 kinase activity can be used as treatment in asthma exacerbations induced by viruses of different origin [1].
In summary, our data showed that inhibition of Pim1 kinase activity reduces viral replication in ALI cultures of PBECs from healthy individuals and moderate-to-severe asthmatic volunteers by enhancing the activity of STAT-1, resulting in an augmented interferon-induced anti-viral response. Further studies focusing on the mechanism of the enhanced interferon response upon inhibition of Pim1 kinase activity should establish our findings and reveal the clinical relevance of inhibition of Pim1 kinase activity as novel therapeutic treatment in virally induced asthma exacerbations.
Acknowledgements
The authors would like to thank Juerg Schwaller (University Children's Hospital of Basel (UKBB), Switzerland) and Andrew Kraft (Medical University of South Carolina, Charleston, SC, USA) for providing the Pim1 inhibitors K00135 and SMI-4a, respectively, and Robert Ridley and Graham Berreen (both University Hospital Southampton, Southampton, UK) for the technical assistance with the ALI PBEC cultures.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This work was supported by ERS Fellowship LTRF 2013–2135 to Maaike de Vries, a shared University of Groningen and University of Southampton PhD studentship (GUIDE PhD studentship to Maaike de Vries) and Medical Research Council (UK) grant number G0900453. Funding information for this article has been deposited with FundRef.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received January 30, 2015.
- Accepted December 9, 2015.
- Copyright ©ERS 2016