





Abstract
The causes underlying the self-perpetuating nature of idiopathic pulmonary fibrosis (IPF), a progressive and usually lethal disease, remain unknown. We hypothesised that alveolar soluble annexin V contributes to lung fibrosis, based on the observation that human IPF bronchoalveolar lavage fluid (BALF) containing high annexin V levels promoted fibroblast involvement in alveolar epithelial wound healing that was reduced when annexin V was depleted from the BALF.
Conditioned medium from annexin V-treated alveolar epithelial type 2 cells (AEC2), but not annexin V per se, induced proliferation of human fibroblasts and contained pro-fibrotic, IPF-associated proteins, as well as pro-inflammatory cytokines that were found to correlate tightly (r>0.95) with annexin V levels in human BALF. ErbB2 receptor tyrosine kinase in AECs was activated by annexin V, and blockade reduced the fibrotic potential of annexin V-treated AEC-conditioned medium. In vivo, aerosol delivery of annexin V to mouse lung induced inflammation, fibrosis and increased hydroxyproline, with activation of Wnt, transforming growth factor-β, mitogen-activated protein kinase and nuclear factor-κB signalling pathways, as seen in IPF.
Chronically increased alveolar annexin V levels, as reflected in increased IPF BALF levels, may contribute to the progression of IPF by inducing the release of pro-fibrotic mediators.
Abstract
Soluble annexin V, found at increased levels in BALF from IPF patients, promotes inflammation and fibrosis in mouse lung http://ow.ly/MHobz
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most lethal interstitial lung disease, with few effective treatment options. The prevalence and mortality of IPF is rising and increases markedly with age [1]. A growing body of evidence indicates that it is the result of multiple cycles of alveolar epithelial type 2 cell (AEC2) injury and activation [2], and biomarkers of AEC2 injury are usually detected in the plasma of IPF patients [3]. Chronic AEC2 damage and subsequent expansion of the fibroblast/myofibroblast population and extracellular matrix accumulation lead to relentless disease progression [4], yet the self-perpetuating nature of IPF has remained an enigma.
Annexins are a multigene family of unique membrane binding proteins that bind to phospholipid in a calcium-dependent manner. Annexin V is found consistently in bodily fluids, suggesting an extracellular role [5]. It is best known as a marker of apoptotic cells, due to its high affinity for phosphatidylserine exposed by damage [6]. Annexin V's known biological activities are based on phosphatidylserine binding and include membrane damage repair [7], anticoagulant activity [8] and modulation of protein kinase C and phospholipase A2 [9, 10]. Annexin V directly interacts with the intracellular domain of the vascular endothelial growth factor (VEGF) receptor-2, and thus may function as a signalling protein in cells that express this receptor [11].
Annexin V is co-secreted with surfactant [12], and can be detected in human bronchoalveolar lavage fluids (BALF) from control patients at levels of ≤20 ng·mL−1 and at significantly higher levels in BALF from IPF patients. This observation led to the novel hypothesis that increased levels of soluble annexin V in the alveolus can play an active, detrimental role in alveolar epithelial wound healing. Herein, we determine the effects of elevated annexin V levels on alveolar epithelium in vivo and in vitro, and confirm that elevated alveolar annexin V in vivo results in lung inflammation and fibrosis, while in vitro, annexin V stimulates the release of pro-fibrotic proteins and cytokines from AECs that are also detected in the BALF of IPF patients.
Materials and methods
Human BALF samples
Anonymous coded samples of discarded human cell-free BALF [13] were obtained from Moises Selman (Interstitial Lung Diseases Program, National Institute of Respiratory Diseases (INER), Mexico City, Mexico), detailed in the online supplementary material.
ELISA kits
Annexin V ELISA kits were obtained from American Diagnostica (Lexington, MA, USA). Mouse connective tissue growth factor (CTGF) ELISA kits were from Biotang (Lexington, MA, USA).
Removal of annexin V from BALF by immunoprecipitation
Annexin V was removed from BALF samples using 20 μL (4 μg) of anti-annexin V (sc-8300; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or 4 μg of rabbit IgG as a control (further details in the online supplementary material).
Cultured cells
Primary cultures of AEC2 were obtained from 250–300 g male rats using the method of Gonzalez and Dobbs [14].
Murine lung epithelial (MLE) 15 AECs [15] were kindly provided by Jeffrey Whitsett (Cincinnati Children's Hospital, Cincinnati, OH, USA).
Human fibroblast cell lines CCD-8Lu (ATCC CCL-201) and WI-38 (ATCC CCL-75) were obtained from ATCC (Manassas, VA, USA).
In vitro alveolar epithelial wound model
Freshly isolated rat AEC2 were plated densely in 24-well plates, and damaged the next day, when confluent, by scraping with a pipette tip. The cells were washed twice to remove debris, and incubated for 24 h with the substance of interest before fixation and vimentin immunostaining. Human BALF was added, diluted 1:1 in DMEM/F12 medium with fetal bovine serum at a final concentration of 10%.
In vivo tracheal administration of annexin V by aerosol
Human recombinant annexin V (eBioscience, San Diego, CA, USA) which has been used in vivo studies [16] was screened for endotoxin and instilled into mouse lung using a MicroSprayer aerosoliser (Penn-Century, Philadelphia, PA, USA) (see online supplementary material for details). Doses were based on annexin V recovered from human IPF BALF, corrected for the dilution of epithelial lining fluid in human BALF and the relative volumes of mouse versus human lung [17–19]. Bleomycin 1.5 U·kg−1 was administered intratracheally to a cohort of animals as a positive control for lung fibrosis. Animal work was performed in compliance with an institutional animal care and use committee-approved protocol.
Recombinant annexin V
Human recombinant annexin V was obtained from two sources: eBioscience and AbD Serotec/BioRad (Raleigh, NC, USA). Each batch of annexin V was tested for endotoxin levels using an assay kit from Genscript (Piscataway, NJ, USA), and was <0.04 endotoxin units (EU)·mL−1 for all batches tested. The 150 μL aerosol administered to each mouse contained <0.006 EU, 4% of the maximum acceptable endotoxin dose that can be administered to a 30-g mouse [20].
Collagen measurement
Hydroxyproline was measured in hydrolysates of the entire lung using a kit from BioVision (Milipitas, CA, USA). Lung sections were stained with celestine blue, haematoxylin and 0.1% Sirius red in saturated picric acid [21]. Soluble collagen was measured in conditioned medium using a quantitative picro-Sirius red assay (online supplementary material).
Proteomic screening
Proteins were separated on a two-dimensional gel and in-gel digested with trypsin [22]. The resulting peptides were cleaned using C18 OmixTips (Varian, Palo Alto, CA, USA) and analysed using an Eksigent nanoLC-2D (SCIEX, Framingham, MA, USA) coupled to an Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA, USA) [23]. Proteins were identified from tandem mass spectra using complementary protein identification software systems, BioWorks (Thermo) and Scaffold (Proteome Software, Portland, OR, USA) as described by Sadygov et al. [24].
Cytokine assay
MILLIPLEX MAP cytokine/chemokine magnetic bead kits (human and mouse) were provided by Millipore (Billerica, MA, USA) and used according to manufacturer's instructions. Analysis was provided by the company using proprietary software (Milliplex Analyst).
Western blot
Western blot analysis was performed as previously described [25]. Lung lysates were made using the entire lung.
Antibodies
Signalling proteins plus their inactive forms were as follows. Extracellular signal-regulated kinase, β-catenin, Smad2, IKBa, Akt and ErbB2 (Cell Signaling Technology, Danvers, MA, USA); proliferating cell nuclear antigen (PCNA), vimentin (for mouse) and annexin V (Santa Cruz Biotechnology); vimentin (for rat) and smooth muscle actin (SMA) (Sigma, St Louis, MO, USA); and β-actin (MP Biomedicals, Solon, OH, USA).
Receptor tyrosine kinase protein array
Mouse receptor tyrosine kinase (RTK) arrays were obtained from R&D (Minneapolis, MN, USA).
Mouse ErbB2 silencing RNA
ON-TARGETplus silencing (si)RNA and control RNA was obtained from GE Dharmacon (Lafayette, CO, USA) and transfected into cells at a final concentration of 10 nM, using Lipofectamine RNAiMAX (Invitrogen/Life Technologies, Grand Island, NY, USA).
Lung imaging
High-resolution micro-computed tomography (CT) scanning of lungs using a SkyScan 1172 Imager (Bruker, Brussels, Belgium) was performed ex vivo, due to the time the scan required (>4 h). Mouse lungs were inflated under 25 cmH2O pressure before imaging.
Data analysis
A t-test was used to compare treated versus untreated groups. For multiple comparisons, ANOVA was used, followed by the appropriate post hoc test as dictated by the normality and distribution of the data. All statistical analyses were performed using GraphPad Prism software (La Jolla, CA, USA). Image J software (National Institutes of Health) was used to calculate Western blot band density. Data represent three individual experiments, unless otherwise stated, and are presented as mean±sem, with p≤0.05 considered significant.
Results
BALF samples from patients with interstitial lung disease promote fibroblast growth in vitro
Primary rat AEC2 cultures, which in our hands reproducibly contain 2–5% of fibroblasts after attachment (fig. 1a), provide an efficient biological screen for monitoring fibroblast involvement in alveolar epithelial wound healing, in response to potential fibrogenic stimuli. Cell-free BALF was obtained from control patients and patients with confirmed lung disease (hypersensitivity pneumonitis, auto-immune disease and IPF), and analysed for pro-fibrotic alveolar epithelial wound healing in a blinded fashion in order to discriminate between the groups. In vitro damaged rat AEC2 treated for 24 h with BALF from control patients (i.e. without interstitial lung disease) show predominantly epithelial growth within the wound, as seen by characteristic morphology and lack of vimentin staining. In contrast, BALF from IPF and autoimmune patients results in predominantly vimentin-staining cells within the wound. BALF from hypersensitivity pneumonitis patients promoted both fibroblast and epithelial growth. α-SMA (myofibroblast) staining was not significantly different between the BALF-treated groups (data not shown), suggesting that increased vimentin staining represented growth of fibroblasts already present in the culture, rather than epithelial–mesenchymal transition (EMT).

Bronchoalveolar lavage fluid (BALF) from idiopathic pulmonary fibrosis (IPF) patients promotes fibroblast growth in alveolar epithelial wounds. Vimentin-stained primary cultures of rat alveolar epithelial type 2 cells (AEC2) containing ∼2–5% fibroblasts before damage, and after damage plus 24 h exposure to BALF from a) control subjects; b) hypersensitivity pneumonitis (HP); c) IPF; and d) autoimmune (AI) disease patients; e) undamaged cells. Representative results for each group (n=5) are presented (scale bar=400 μm). Arrows show predominantly AEC growth within the wound in cultures treated with BALF from control and HP patients, while increased vimentin-stained cells are seen in cultures treated with IPF and AI BALFs. f) BALF from IPF patients contains high levels of soluble annexin V. Annexin V levels (quantified by human-specific annexin V ELISA) were higher in BALF from IPF and AI patients, compared with levels in control BALF. Control n=5; IPF, AI and HP n=6. *: p<0.05; **: p<0.01, by ANOVA followed by Tukey's post hoc test. g) Annexin V induces release of cytokines from AECs that correlate with annexin V in human BALF. In vitro-damaged AECs (murine lung epithelial (MLE)-15 cell line, to ensure 100% epithelial phenotype) were incubated for 24 h with or without 50 ng·mL−1 annexin V, and the conditioned medium (CM) was then screened for murine cytokines, using a Millipore Milliplex 32 cytokine multiplex assay (Billerica, MA, USA). Cytokines strongly associated with annexin V in human BALF, interferon-γ-inducible protein (IP)10 and murine homologues of interleukin-8, keratinocyte chemoattractant (KC) and macrophage inflammatory protein (MIP)2 were released from damaged AECs in response to annexin V. Leukaemia inhibitory factor (LIF) was not available on the human cytokine panel used to obtain the human BALF data. *: p<0.05; **: p<0.01; #: p<0.03 versus control.
Annexin V levels are increased in BALF from patients with interstitial lung disease
Proteomic screening was performed to identify novel proteins in pro-fibrotic human BALF (online supplementary table 1). Annexin V was revealed as a protein of interest exclusive to human BALF that promoted fibroblast growth in epithelial wounds, versus BALF that mediated epithelial wound healing. A human-specific ELISA confirmed significantly increased annexin V levels in BALF from patients with IPF and autoimmune disease (fig. 1f). Thus annexin V was chosen for further study since it is secreted by AEC2 with surfactant [12], is associated with damage, and, as yet, has never been implicated in playing a functional role in fibrotic lung disease.
Proteins secreted by alveolar epithelial type 2 cells in response to annexin V
Specific cytokines correlate with annexin V in human BALF
We first determined which cytokines correlated with annexin V levels in human BALF. Multiplex cytokine analysis of six human BALF samples representing a broad range of annexin V levels from <20 ng·mL−1 to 80 ng·mL−1 revealed that five cytokines from a panel of 40 correlated with BALF annexin V concentrations with r≥0.95: GRO/CXCL1 (r=1.0), interferon-γ-inducible protein (IP)10 (r=0.99), interleukin (IL)-8 (r=0.96), vascular endothelium growth factor, VEGF (r=0.96) and monocyte chemoattractant protein-1 (r=0.95).
Annexin V induces release of cytokines from mouse MLE15 AECs that are shown to correlate with annexin V in human BALF
Four cytokines, from a panel of 32, were secreted by cultured MLE15 AECs at levels of >5 pg·mL−1 in response to 24 h treatment with annexin V (fig. 1g), including three of the cytokines that we found to tightly correlate (r>0.95) with soluble annexin V levels in human BALF. Annexin V stimulated release of IP10, keratinocyte chemoattractant (KC) (rodent homologue of GRO-α) and macrophage inflammatory protein (MIP)2 from damaged AECs. Rodents lack a direct homologue of human IL-8, but CXCL1/KC and CXCL2/MIP2 are regarded as functional homologues [26]. Leukaemia inhibitory factor, the fourth cytokine induced by annexin V in mouse AECs, was not available on the human multiplex panel used for the BALF, but is elevated in IPF lung [27].
Removal of annexin V from IPF BALF reduces fibroblast growth in alveolar epithelial wounds
Annexin V was depleted from three IPF BALF samples by immunoprecipitation (51–80 ng·mL−1 before depletion, ≤10 ng·mL−1 after depletion, as confirmed by ELISA), and added to undamaged and freshly damaged rat AEC2 for 24 h. Annexin V depletion reduced vimentin-staining cells within the cultures, and especially within the wounds (fig. 2a). Western blot analysis of culture lysates confirmed the decrease of vimentin-positive cells (fig. 2b).
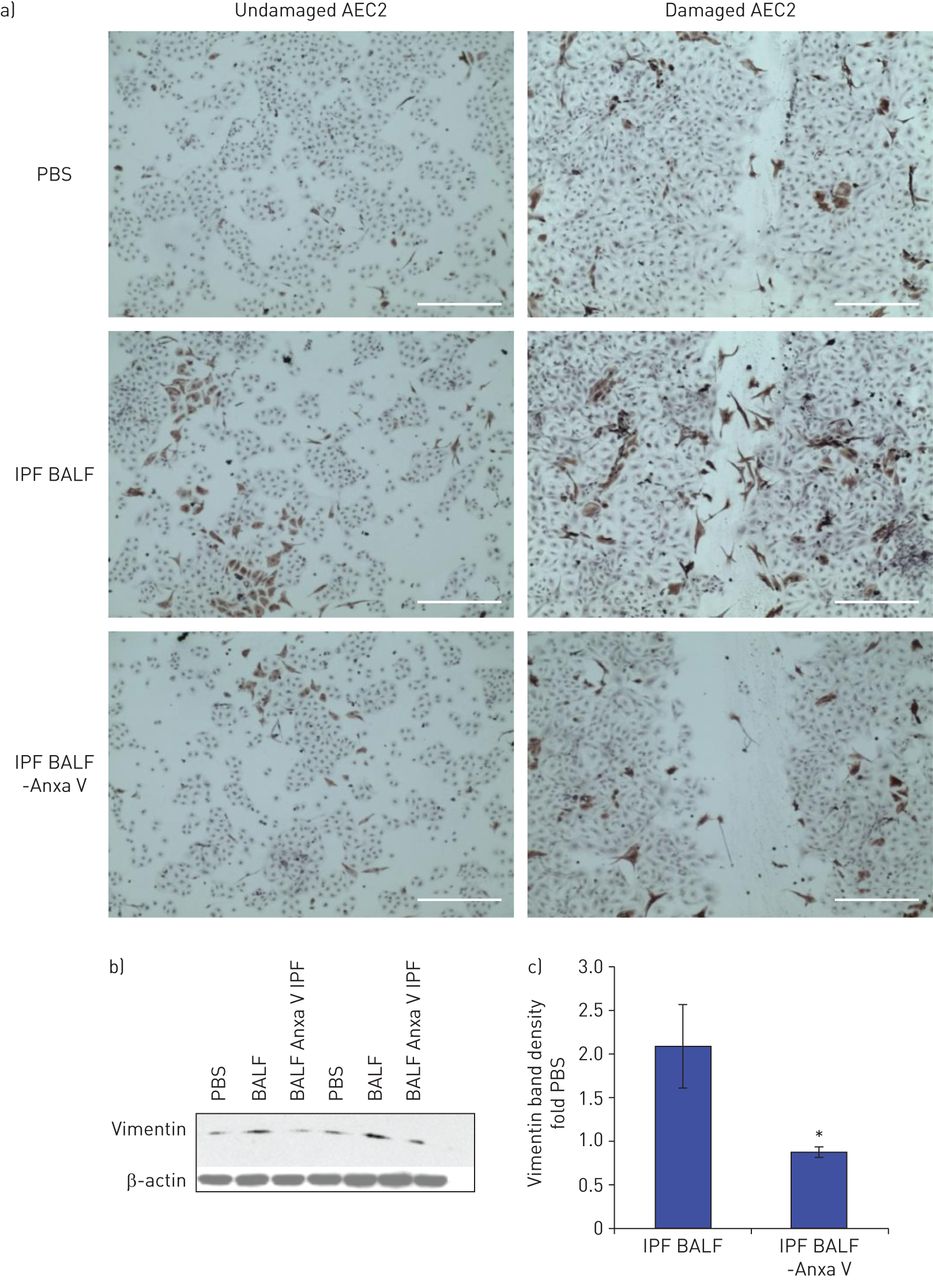
Depletion of annexin V from idiopathic pulmonary fibrosis (IPF) bronchoalveolar lavage fluid (BALF) reduces fibroblast growth in alveolar epithelial wounds. a) Annexin V-depleted (-Anxa V) and undepleted BALF or PBS were added to undamaged and damaged rat AEC2 for 24 h, then the cultures were fixed in 4% paraformaldehyde and stained for vimentin (scale bar=400 μm). The experiment is representative of three individual experiments using different BALF samples. b) Western blotting shows vimentin expression in damaged rat AEC2 treated with two different human IPF BALFs±annexin V depletion. c) Densitometric quantitation of vimentin bands corrected for actin from three Western blots shows less BALF-induced vimentin with annexin-V depletion; n=3; *: p<0.05.
Annexin V added to AEC cultures increases vimentin-staining cells
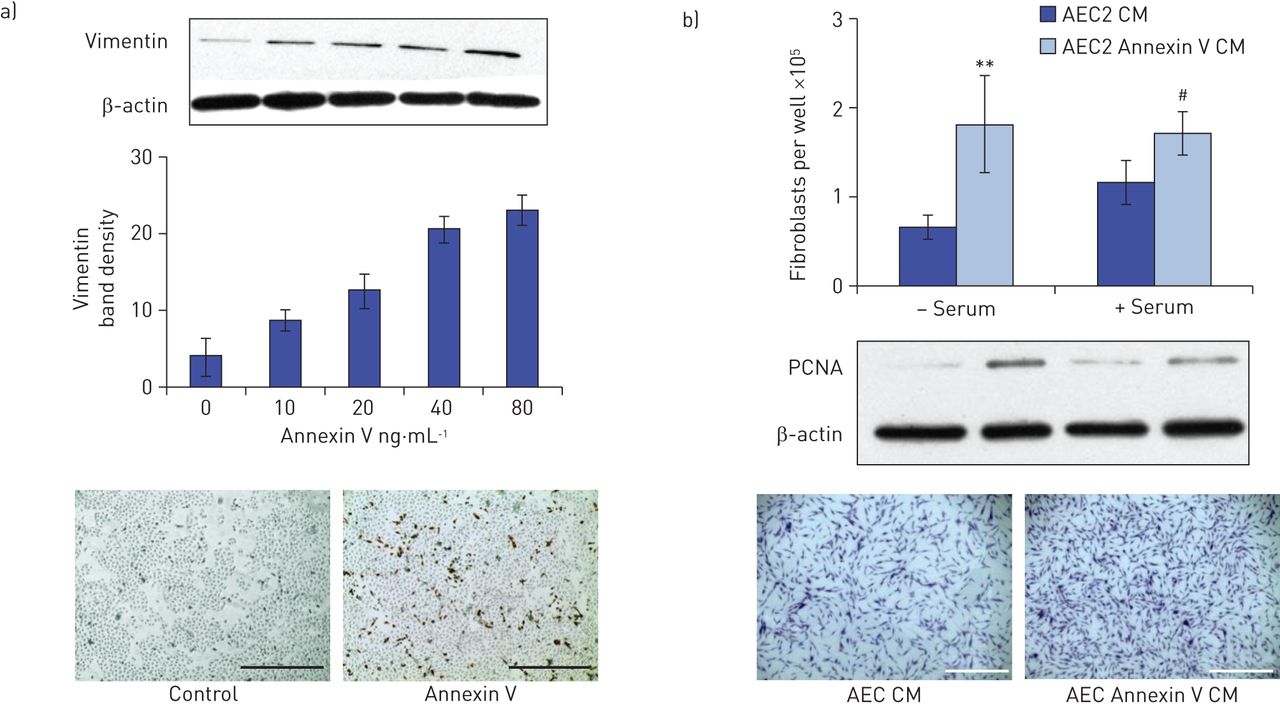
Vimentin expression was induced in primary rat AEC2 cultures treated for 24 h with annexin V in a dose-responsive manner, with maximal expression at 50 ng·mL−1 (fig. 3a). α-SMA expression did not change with annexin V treatment (not shown).

Conditioned medium from annexin V-treated alveolar epithelial type 2 cells (AEC2) promotes fibroblast proliferation. a) Treatment of primary cultures of rat AEC2 with annexin V for 24 h induces a dose-dependent increase in vimentin expression, as shown in the representative Western blot; densitometric analysis of two independent experiments, corrected for β-actin; vimentin staining of control and 50 ng·mL−1 annexin V-treated AEC2 cultures, 10× magnification, scale bar=400 μm. b) Fibroblasts, which did not proliferate in response to annexin V per se (online supplementary fig. 1), were treated for 24 h with conditioned medium (CM) recovered from rat AEC2 that had been treated ±50 ng·mL−1 annexin V, in the presence and absence of serum. After 24 h, the cells were either trypsinised, neutralised and counted, lysed for Western blot analysis of proliferating cell nuclear antigen (PCNA), or stained with crystal violet for visualisation. CM from AEC2 treated with annexin V induced fibroblast proliferation, as measured by cell counts (n=4; **: p<0.01; #: p<0.02 versus control) and PCNA protein expression. Images show crystal violet staining in an experiment where serum was present; scale bar=1000 μm.
Conditioned medium from annexin V-treated AEC2, but not annexin V per se, promotes fibroblast growth
Surprisingly, annexin V did not directly stimulate growth of pure fibroblast cell lines (WI38, NIH3T3 and CCD-8Lu) when added at multiple doses for 24 h, as measured by cell counts and PCNA expression (online supplementary fig. 1). In contrast, conditioned medium from annexin V-treated rat AEC2 induced growth in adult human fibroblast cultures compared with conditioned medium from untreated cells (fig. 3b).
Annexin V stimulates release of pro-fibrotic proteins from AEC2
15 proteins were identified by proteomic screening as exclusive to, or overexpressed in conditioned medium from annexin-treated rat AEC2 (online supplementary tables 2 and 3). Of these, 13 proteins have been associated with IPF, tissue fibrosis, chronic wounding or EMT, including two molecules associated with transforming growth factor (TGF)-β1 signalling, CTGF and plasminogen activator inhibitor (PAI)-1 (table 1).
The two other proteins that were increased by annexin V treatment (phospholipase Cε and eukaryotic translation factor) have no reported association with fibrosis. To validate the screening, CTGF was quantitated by ELISA. Annexin V treatment of MLE15 AECs induced a 1.5-fold increase in secreted CTGF levels versus untreated cells (60.8±1.2 versus 40.9±3.9 pg·mL−1, p<0.004; n=3). Importantly, a proteomic screen of conditioned medium from lung fibroblasts treated for 24 h±annexin V showed that none of the above proteins were secreted by fibroblasts (online supplementary table 4). Interestingly, fibroblasts secreted pro-fibrotic protein collagens 1A1 and 1A2, decorin and fibronectin in response to annexin V. To validate the screening, soluble collagen levels were confirmed to be elevated 1.5±0.06-fold in conditioned medium from annexin V treated fibroblasts versus conditioned medium from controls (p<0.02, n=4). Secretion of collagen was not induced in annexin V-treated MLE15.
Increased alveolar annexin V induces inflammation and collagen deposition in mouse lung
Annexin V solution or vehicle, tested for endotoxin, was administered to 8-week-old C57BL mice by intratracheal aerosolisation, twice per week for 2 weeks, at doses ranging from 7.5 to 75 ng per lung. At 14 days after the last dose of annexin V (28 days after the initial instillation), areas of damage and inflammation were seen in the lungs of annexin V-treated animals (fig. 4a and b), where widely distributed lesions were observed in all lobes. Lungs treated with the low dose (7.5 ng) were not different from control lungs (not shown). Sirius-red staining confirmed increased interstitial collagen deposited around areas of infiltration and along the pleural membrane (fig. 4c and d). Lung hydroxyproline levels were elevated 1.4-fold by annexin V at 28 days (annexin V treated versus control lung 0.25±0.02 versus 0.18±0.02 μg·mg−1 lung; n=4 whole lungs per group). This increase is similar to the 1.6-fold increase seen in bleomycin-treated lung at 28 days post-bleomycin [40]. High-resolution micro-CT scans of annexin-treated lungs 28 days after the first instillation of annexin V showed fibrosis as lace-patterned opacities in trans-axial slices which was present in all lobes, and thickened interlobar and alveolar septa (fig. 4e–g). The major difference detected between the annexin V-treated versus bleomycin-treated lung at 28 days is the persistence of infiltration in the annexin V-treated lung, which is probably due to impaired efferocytosis due to excess annexin V [41]. Annexin V-induced lesions had resolved by 2 months after annexin V treatment ceased, but persistent thickening along the pleural membrane was seen in some animals (not shown).

Intratracheal aerosolised annexin V induces infiltration and collagen deposition in mouse lung. Interstitial inflammation and patchy fibrosis was seen in the lungs of mice that received annexin V by intratracheal aerosol twice per week for 2 weeks to mimic a limited chronic exposure, as seen in haematoxylin and eosin stained lung sections of a) control and b) 75 ng annexin V, 28 days after the initial annexin V dose. 4× magnification; scale bar=500 μm. The infiltrate was predominantly Ly6G-positive cells, as determined by immunostaining (not shown). Lungs treated with a dose reflecting annexin V levels in control human bronchoalveolar lavage fluid (7.5 ng), were not different from control lungs (not shown). Increased collagen was detected in the lungs of d) annexin V-treated versus c) vehicle-treated mice, as patchy areas of Sirius red within the lung interstitium associated with areas of infiltration, and along the pleural membrane (arrows) at 28 days after the initial treatment. 20× magnification; scale bar=100 μm. Hydroxyproline was elevated 1.4-fold in lung hydrolysates from mice treated with 75 ng annexin V 28 days after the initial annexin V treatment (p<0.02, n=4), an increase similar to that induced by bleomycin (1.6-fold) at day 28 post-bleomycin [40]. Micro computed tomography scans of e) control, f) bleomycin-treated lung and g) annexin V-treated lungs. g) Annexin V-treated lung inflated to 20 cm water (75 ng annexin V per lung) performed 28 days after the first annexin V treatment, shows extensive fibrosis, seen as lace-patterned opacities (lower arrow) in trans-axial slice, which was present in all lobes, upper arrow shows thickened intralobar septa; f) bleomycin-treated lung at 28 days (fibrotic phase) of injury serves as positive fibrotic control, and also shows patchy fibrosis (arrow). Despite similarities, the major difference detected between the annexin V-treated versus bleomycin-treated lung at 28 days is the persistence of infiltration in the annexin V-treated lung.
Annexin V activates signalling pathways in AECs and lung
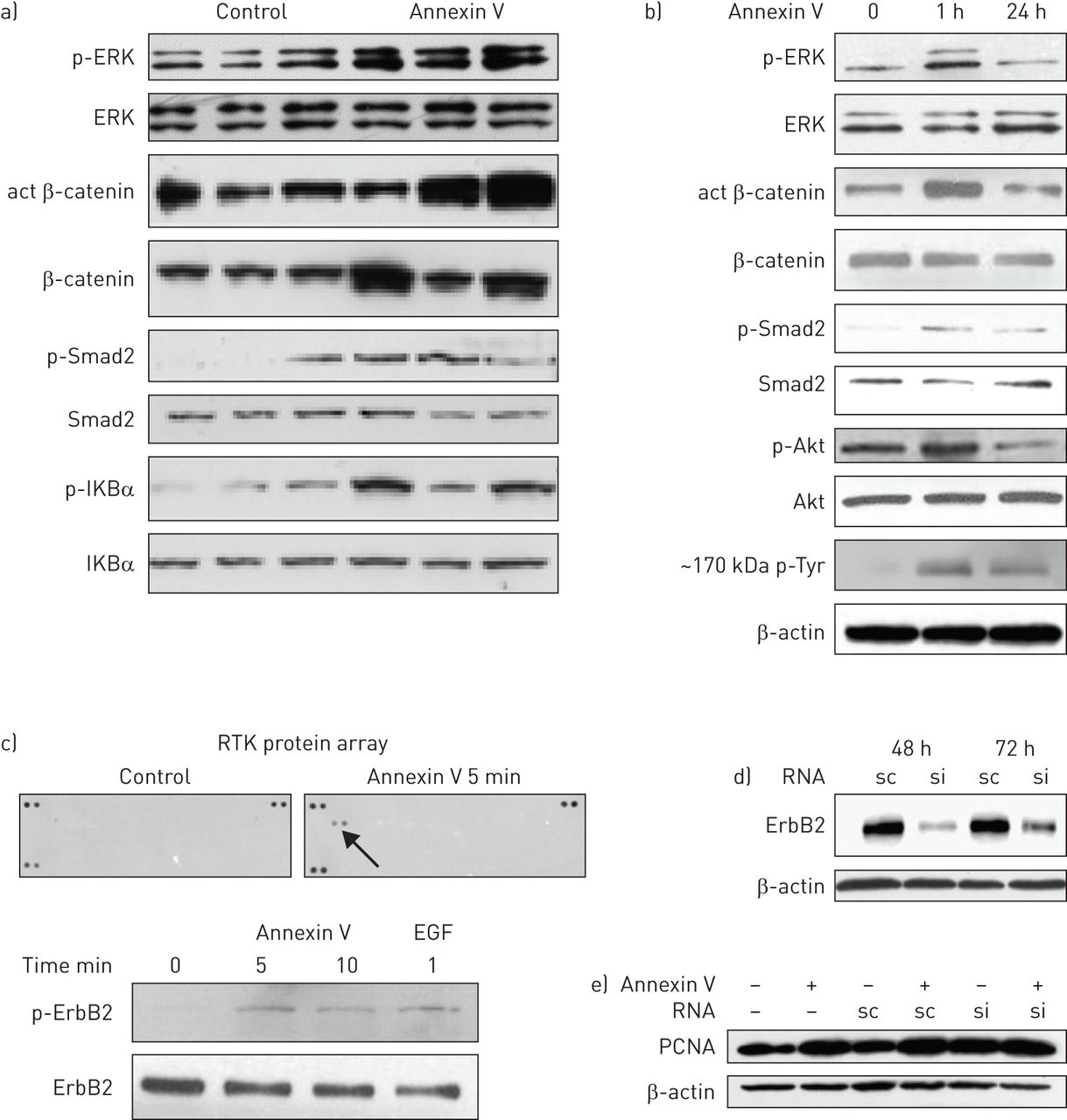
Wnt, TGF-β, nuclear factor (NF)κB and mitogen-activated protein kinase (MAPK) signalling pathways were activated in 75 ng annexin V-treated lungs 28 days post-treatment (fig. 5a), as seen in the lungs of IPF patients [42]. The Wnt, MAPK, TGF-β and PI3K/Akt pathways were activated within 1 h of annexin V exposure to rat AEC2, as seen in figure 5b. Blotting for phosphotyrosine showed induction of a high-molecular-weight protein (∼150–200 kDa) with short- and long-term annexin V treatment. MLE15 AECs responded similarly (data not shown). Activation of the NF-κB pathway was also detected, but only after 24 h exposure, indicating a secondary response (data not shown).
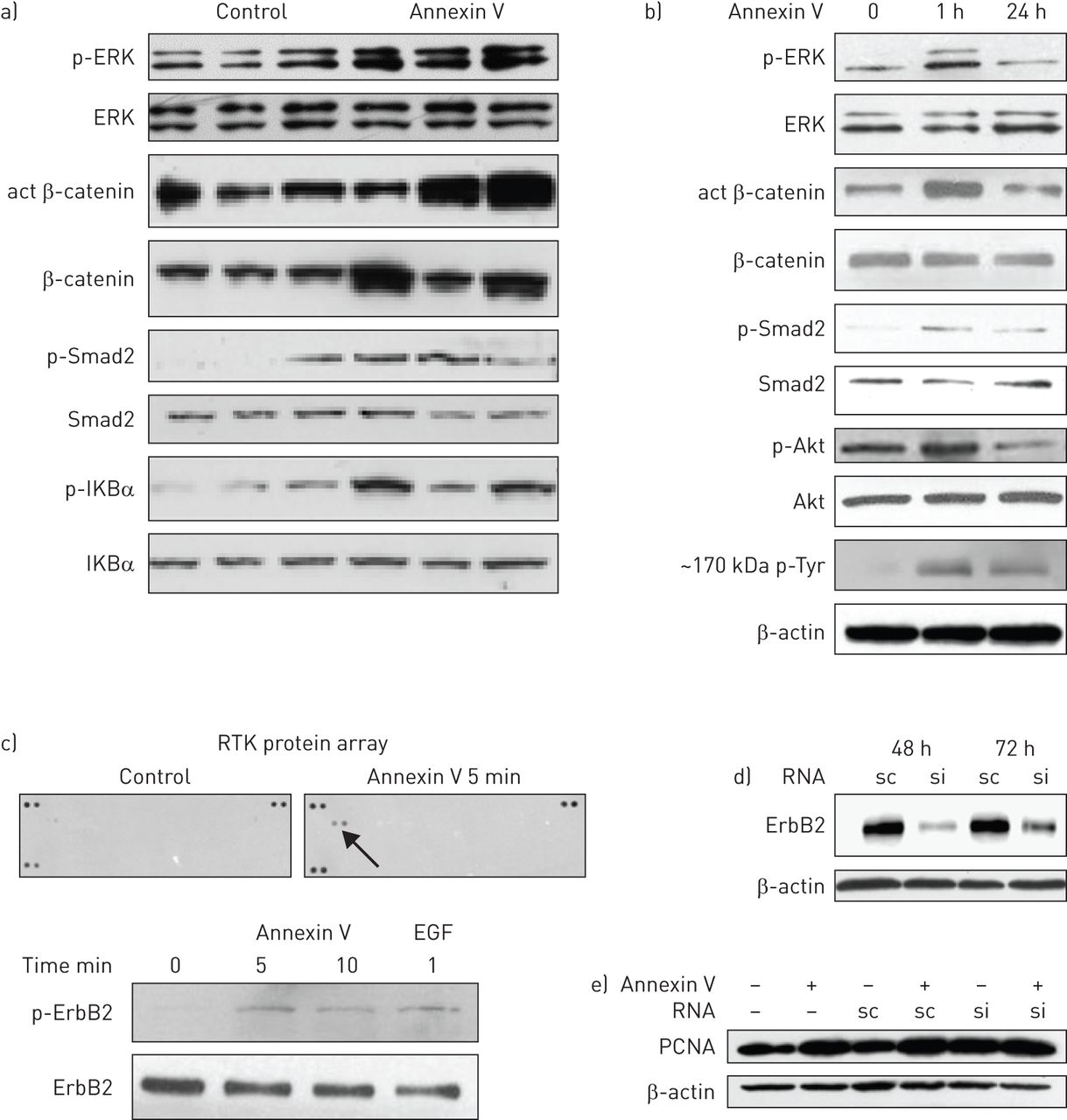
Annexin V activates signalling pathways in alveolar epithelial cells (AECs) and in the lung. a) Lysates of three control and three annexin V-treated mouse lungs, 28 days after treatment with annexin V (75 ng per lung), were analysed by Western blotting for the key proteins that are activated in major signalling pathways in idiopathic pulmonary fibrosis. Due to the patchy nature of annexin V-mediated lung injury, the whole lung was used to ensure representative data. Wnt, transforming growth factor (TGF)-β, nulcear factor κB and mitogen-activated protein kinase (MAPK) pathways were activated by annexin V treatment, as seen by the increased active (act) β-catenin, p-Smad2, p-IKBa and p-extracellular signal-regulated kinase (ERK), respectively, versus their nonphosphorylated forms. b) Active signalling proteins from MAPK, Wnt, TGF-β and PI3K/Akt pathways and a high-molecular-weight (∼150–200 kDa) tyrosine phosphorylated protein were detected after 1 h of annexin V exposure to rat AEC2. The same results were seen when the murine lung epithelial (MLE)15 AEC cell line was used (data not shown). c) ErbB2 tyrosine kinase phosphorylation/activity was detected by receptor tyrosine kinase (RTK) antibody array in lysates of MLE15 AECs that had been treated for 5 min with 50 ng·mL−1 annexin V in the absence of serum. Activation of ErbB2 by annexin V was confirmed by Western blotting using epidermal growth factor (EGF) 10 ng·mL−1 for 1 min as a positive control. d) MLE15 AECs were transfected with ErbB2 silencing (si)RNA or scrambled (sc)RNA (10 nM) using Lipofectamine RNAiMAX (Invitrogen/Life Technologies, Grand Island, NY, USA), then lysed for Western blotting at 48 h and 72 h post-transfection to confirm silencing. e) Conditioned medium from MLE15 AECs that had been cultured ± annexin V ± ErbB2 silencing was added to nonconfluent wells of fibroblasts for 24 h, then the cells were lysed for Western blot of proliferating cell nuclear antigen (PCNA). The blot is representative of three individual silencing experiments, which showed that the increase in fibroblast PCNA induced by AEC–annexin V conditioned medium was abolished when AEC2 ErbB2 was silenced (quantitation, by densitometry of PCNA bands and cell counts is presented in online supplementary fig. 2).
ErbB2 phosphorylation in AECs is induced by annexin V
A protein array of 39 mouse RTK antibodies was used to identify potential RTKs activated in MLE15 AECs by annexin V. Unexpectedly, ErbB2, a member of the epidermal growth factor receptor family, was the only RTK whose activation was detected in AECs within 5 min of annexin V treatment, which was confirmed by Western blotting (fig. 5c).
Silencing ErbB2 or inhibition of ErbB tyrosine kinase activity reduces annexin V-mediated secretion of pro-fibrotic proteins from AECs
ErbB2 was effectively silenced in MLE AECs using 48 h treatment with siRNA versus scrambled (sc)RNA (fig. 5d). Conditioned medium from MLE15 AECs treated with ErbB2 siRNA/annexin, when added to adult human fibroblasts for 24 h, induced less fibroblast growth than conditioned medium from scRNA/annexin-treated AECs, as measured by PCNA expression (fig. 5e) and cell counts (online supplementary fig. 2, which also shows quantitation of PCNA band data). Conditioned medium from MLE15 AECs that had been treated with annexin V in the presence of the cell permeable irreversible inhibitor of ErbB tyrosine kinase activity, PD 168393 (10 μM), also induced less fibroblast growth than conditioned medium from annexin V-treated AECs, as measured by PCNA expression (fig. 6a) and cell growth (fig. 6b). The 1.5-fold increase in secreted CTGF that was induced by 24 h annexin treatment of AECs was reduced by both ErbB kinase inhibition and silencing ErbB2 (fig. 6c). These data obtained from two different blocking strategies suggest that annexin V-mediated ErbB activation in AECs is upstream of the release of pro-fibrotic factors, including CTGF. Of the three cytokines released from annexin-treated AECs that were found to correlate with annexin V in human BALF (IP10, KC and MIP-2), only IP10 was found to be downstream of ErbB RTK activation (online supplementary fig. 3), suggesting that annexin-mediated release of KC and MIP-2 was occurring by a different mechanism.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ErbB receptor tyrosine kinase inhibition in alveolar epithelial cells (AECs) reduces annexin V-mediated secretion of pro-fibrotic proteins. a) Conditioned medium (CM), collected from murine lung epithelial (MLE)15 AECs that had been preincubated with 10 μM ErbB receptor tyrosine kinase inhibitor PD168393 or dimethyl sulfoxide (inhibitor diluent) for 2 h before the addition of annexin V (50 ng·mL−1) for 24 h, was added to nonconfluent cultures of fibroblasts for 24 h. Wells were lysed for Western blotting of PCNA to compare cell growth. conditioned medium from AEC treated with annexin V induced fibroblast growth as expected, as shown by induction of proliferating cell nuclear antigen (PCNA). In contrast, CM from AECs preincubated for 2 h with PD168393 before the addition of annexin did not induce fibroblast growth versus CM from AECs treated with PD168393 alone. b) Parallel wells treated as in a) were fixed and crystal violet-stained. c) Annexin V-mediated connective tissue growth factor (CTGF) secretion from MLE15 AECs, measured by ELISA, is reduced by ErbB receptor tyrosine kinase inhibition (n=4) or ErbB2 silencing (si)RNA (n=3), by ANOVA followed by Tukey's post hoc test. ERK: extracellular signal-regulated kinase; scRNA: scrambled RNA. *: p<0.05.
Discussion
Little is known about the origin and function of annexin V in body fluids [5]. The homeostatic levels (≤20 ng·mL−1) of soluble annexin V that we detected in normal human BALF are likely to originate from normal alveolar cell turnover and secretion by AEC2 along with surfactant. We detected low levels of annexin V in the culture medium of undamaged rat AEC2 (∼2 ng·10–6 cells), which increase after in vitro damage (data not shown). Cultured rat AEC2 and mouse MLE15 cells that were treated with annexin V at the elevated levels found in the BALF of IPF patients released pro-fibrotic proteins, as well as pro-inflammatory cytokines that are also detected in IPF BALF and correlate tightly (r≥0.95) with annexin V levels. Human lung fibroblasts also responded to annexin V by secretion of matrix proteins associated with fibrosis. If AEC2 in the human IPF lung respond to annexin V as seen in vitro with rodent AECs, the following provisional scenario for disease progression is suggested: damaged and/or aberrantly activated AEC2 [3] release increased levels of soluble annexin V, which promotes autocrine and paracrine secretion of pro-fibrotic proteins. In addition, phosphatidylserine-mediated macrophage efferocytosis, which clears damaged cells from the alveolus, is impeded by excess annexin V, which effectively masks the exposed phosphatidylserine on damaged cells that is the signal for their removal [42]. Therefore, the infiltration induced by annexin V-mediated cytokines would be expected to clear less efficiently, resulting in further cell damage and annexin V release, thus perpetuating the cycle of annexin V release, infiltration and fibrosis. The persistence of lung infiltrates in mice treated with annexin V is consistent with this speculation.
The lungs of annexin V-treated mice showed the hallmarks of early fibrotic lung disease, including increased fibrosis and inflammation, with elevated hydroxyproline levels and increased Wnt, MAPK and TGF-β signalling activity. The activation of TGF-β signalling observed in the annexin V-injured mice correlates with the phosphorylation of Smad2 and with the increase of CTGF and PAI-1 (downstream mediators of TGF-β) revealed in AEC2 stimulated with annexin V. TGF-β, secreted and activated by damaged AEC2 plays a critical role in the development of IPF by promoting EMT, collagen synthesis, and fibroblast proliferation [4]. Activation of the Wnt signalling pathway was also detected in annexin V-treated mouse lungs, as is seen in IPF, and AEC responded to annexin V treatment by activation of β-catenin and by secretion of secreted frizzled-related protein (sFRP)-1, a Wnt target protein that is elevated in IPF lungs [40]. The MAPK signalling pathway, which is activated in IPF lungs and exhibits significant crosstalk with both TGF-β and Wnt pathways, was also activated in annexin V-treated lungs as well as in annexin V-treated rat AECs.
ErbB2 tyrosine kinase activity was induced in AECs within 5 min of exposure to annexin V, and blockade using two different strategies was effective in reducing the fibroblast-stimulatory activity of annexin V-conditioned AEC medium. Specifically, annexin V-mediated CTGF release from AECs was found to be downstream of ErbB2 activation. Induction of CTGF following ErbB2 activation has previously been reported in fibroblasts [43]. It remains to be determined whether annexin V itself binds directly to ErbB2 on AECs, and whether other annexin family members can elicit similar responses from AECs.
We speculate that reduction of excessive alveolar annexin V to homeostatic levels has the potential to moderate multiple pathways and downstream targets in lung that have already been implicated in fibrosis, yet whose individual blockade has proved ineffective in clinical trials.
Acknowledgements
The authors would like to thank Gevorg Karapetyan and the members of the Children's Hospital Los Angeles Small Animal Imaging Core (Los Angeles, CA, USA) for their invaluable help with the lung micro-computed tomography scans.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
Support statement: This study was funded by the United States Department of Health and Human Services, National Institutes of Health (NIH) (RO1 HL44060 to D. Warburton; HL068597 and HL109932 to W. Shi; R01 DK095004 to M.R. Frey; and UO1 HL122681 to W. Shi, R. Moats and D. Warburton), the American Cancer Society (Research Scholar Grant 124678 to M.R. Frey), the Pasadena Guild of Children's Hospital Los Angeles Endowment and the Garland Foundation and the Webb foundation (to D. Warburton). Funding information for this article has been deposited with FundRef.
- Received January 7, 2015.
- Accepted April 25, 2015.
- Copyright ©ERS 2015
References










