





Abstract
Pulmonary arterial hypertension (PAH) is a devastating cardiopulmonary disorder with various origins. All forms of PAH share a common pulmonary arteriopathy characterised by vasoconstriction, remodelling of the pre-capillary pulmonary vessel wall, and in situ thrombosis. Although the pathogenesis of PAH is recognised as a complex and multifactorial process, there is growing evidence that potassium channels dysfunction in pulmonary artery smooth muscle cells is a hallmark of PAH. Besides regulating many physiological functions, reduced potassium channels expression and/or activity have significant effects on PAH establishment and progression. This review describes the molecular mechanisms and physiological consequences of potassium channel modulation. Special emphasis is placed on KCNA5 (Kv1.5) and KCNK3 (TASK1), which are considered to play a central role in determining pulmonary vascular tone and may represent attractive therapeutic targets in the treatment of PAH.
Abstract
Comprehensive overview of the roles of potassium channels in the pathogenesis of pulmonary arterial hypertension http://ow.ly/QyBzX
Introduction
Pulmonary hypertension (PH) encompasses a heterogeneous group of diseases, haemodynamically defined by a mean pulmonary artery pressure >25 mmHg, that share histological similarities but differ in pathophysiology and prognosis. The current classification identifies five groups [1]. Among these, the pulmonary arterial hypertension (PAH) group includes idiopathic and hereditary entities, PAH associated with connective tissue disorders, congenital heart diseases or HIV infection as well as persistent PH of the newborn. PAH is characterised by proliferation and remodelling of the small pulmonary arteries, leading to increased pulmonary vascular resistance, subsequent right ventricular hypertrophy and ultimately death. It is currently accepted that endothelial cell dysfunction, originating from genetic defects, hypoxia, shear stress or inflammation, promotes the release of vasoconstrictors and mitogens that in turn trigger pulmonary artery smooth muscle cells (PASMCs) contraction, proliferation and resistance to apoptosis [2, 3]. Although pathogenesis of PAH is recognised as a complex and multifactorial process, numerous data has accumulated demonstrating the significant role of potassium (K+) channels in the cellular mechanisms underlying abnormal PASMC behaviour. In regulating potassium flow across the membrane and subsequent modulation of cytoplasmic free calcium concentration ([Ca2+]cyt), potassium channels control a plethora of biological functions. This review summarises current knowledge regarding their involvement in the development of PAH.
Overview of potassium channels
Most of our knowledge regarding regulation of K+ channel expression and function in PAH remains somewhat indirect and almost exclusively relies on the interpretation of electrophysiological data obtained by using pharmacological K+ channel modulators. K+ channels are transmembrane proteins connecting the cell interior to the extracellular environment by forming a pore in the cytoplasmic membrane. They are the largest family of membrane ion channels and are categorised into three major groups based on their structure as well as their electrophysiological and pharmacological properties: 2TM, 4TM, and 6TM, corresponding to the number of transmembrane (TM) spanning regions possessed by each of the α-subunits that make up the channel. The first group consists of K+ channels with two TM domains in each of their α-subunits, also known as inward rectifier K+ (KIR) channels. In contrast to the 2TM and 6TM families, 4TM channels possess two rather one pore-forming domain per subunit. 4TM channels are often referred as two-pore domain K+ channels or K2P. Designated leak or background channels, they are mostly voltage-independent and contribute to setting the resting membrane potential. They respond to various physical and chemical stimuli such as mechanical stretch, pH and volatile anaesthetics. The 6TM channels are the largest class of K+ channels and are subdivided into two types: calcium-activated K+ (KCa) channels and voltage-dependent K+ (KV) channels. KCa channels are subdivided into BK, IK, and SK channels according to conductance (big, intermediate, and small). Additional K+ channel diversity is generated from alternative RNA splicing, post-translational modifications, the presence of both homomeric and heteromeric assemblies of the various α-subunits, and the existence of accessory β-subunits modulating the functional properties of the channel. K+ channels can be regulated at multiple levels by factors controlling channel expression, biosynthesis, trafficking by specialised chaperone proteins, insertion into the cell membrane, recycling, degradation, and activity. Alterations in these mechanisms can modify K+ channel density and activity at the cell surface and therefore profoundly impact cell homeostasis.
Potassium channels in regulation of cell proliferation and cell death
Tonically active in vascular smooth muscle cells, K+ channels are the major regulator of resting membrane potential. Under normoxic conditions, an outward K+ current flows through these channels. This current maintains a resting membrane potential in the range of −50– −60 mV and blocks calcium entry through voltage-activated Ca2+ channels. Inhibition of K+ channel activity results in accumulation of positively charged K+ ions within the cell. This renders the interior of the cell more positive resulting in membrane depolarisation and activation of voltage-activated Ca2+ channels, leading to an increase in [Ca2+]cyt and vasoconstriction (fig. 1). The rise in [Ca2+]cyt can be caused by two closely related mechanisms: 1) influx of Ca2+ from extracellular fluid, and 2) release of Ca2+ from intracellular storage sites such as the sarcoplasmic reticulum. Any prolonged variations in intracellular K+ and Ca2+ concentrations directly influence cell survival and proliferation (fig. 1). By governing K+ movement and intracellular osmolality that drives water flow across cell membrane, K+ channels are crucial regulators for cell volume. In PASMCs, increased K+ efflux, either by transient transfection of a KV channel or by a K+ channel activator, enhances cell death, a conserved mechanism characterised by cell shrinkage, chromatin condensation, nuclear fragmentation and apoptotic body formation [4–6]. Conversely, inhibition of K+ efflux attenuates PASMC apoptosis [7]. The critical role of potassium channels in apoptosis has been substantiated in a study in which the anti-apoptotic protein Bcl-2 was overexpressed in rat PASMCs. Sustained expression of Bcl-2 caused a marked diminution of the KV1.1, KV1.5 and KV2.1 transcript levels that subsequently decrease whole cell KV current. Increases in KV current preceding PASMC apoptosis induced by staurosporine were suppressed by overexpression of Bcl-2, indicating that KV channel activation-mediated apoptotic volume decrease is an early prerequisite for apoptosis [8]. In addition, the consequences of high intracellular calcium levels on PASMC growth are well documented enforcing a cellular phenotype that is resistant to apoptosis and promoting cell proliferation [9]. Based on the physiological roles and properties of K+ channels in PASMCs, the implication of K+ channels in PAH has been the subject of intensive research.
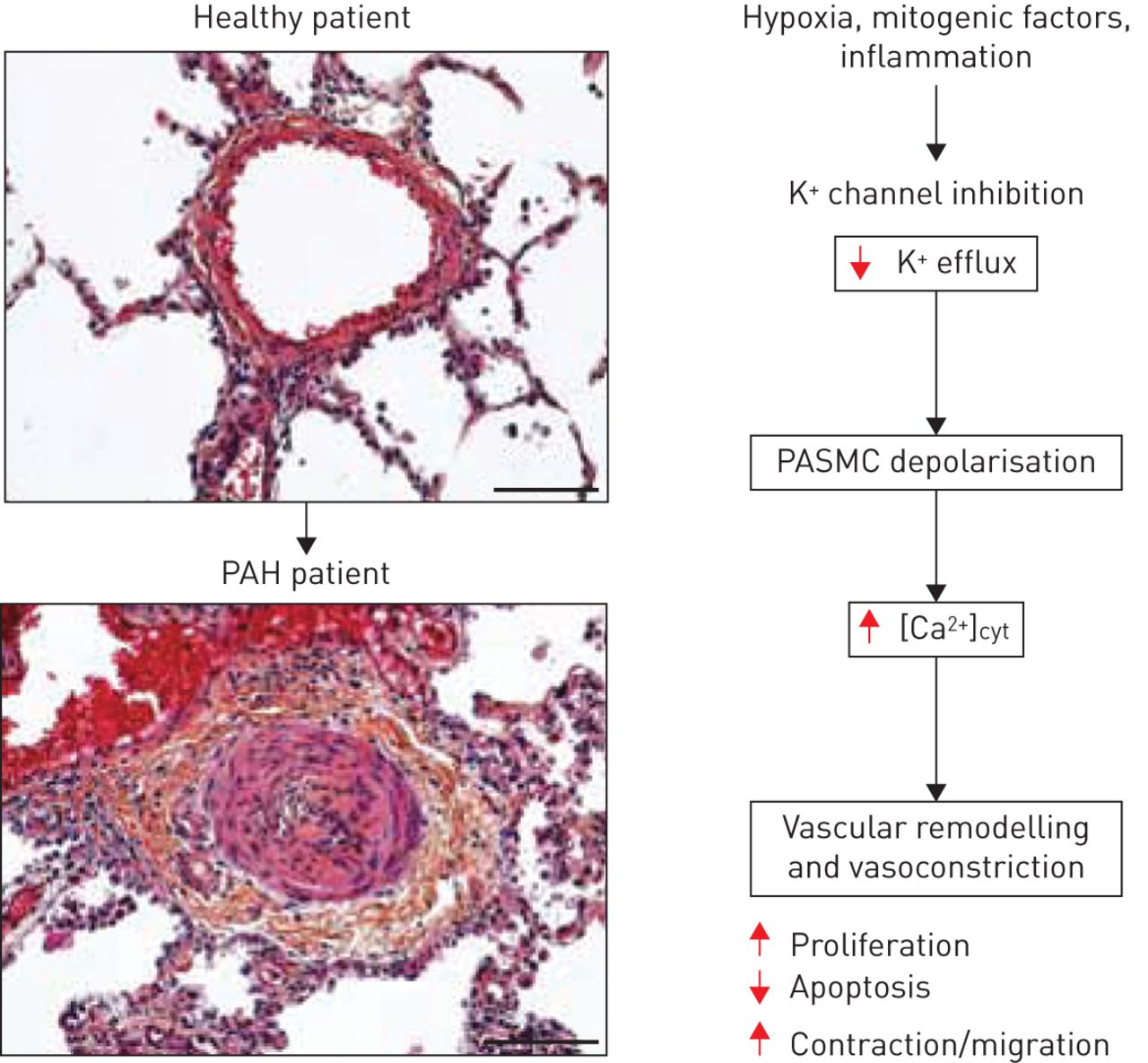
K+ channel inhibition in pulmonary artery smooth muscle cells (PASMCs) leads to membrane depolarisation. This causes an increase in intracellular calcium levels that promotes cell proliferation and contraction as well as resistance to apoptosis and triggers pulmonary vasoconstriction and excessive vascular remodelling. Healthy patient scale bar=100 μm; pulmonary arterial hypertension (PAH) patient scale bar=100 μm. [Ca2+]cyt: cytoplasmic free calcium concentration.
KV channel dysfunction as a contributing factor in the development of PAH
KV channels are tetramers of α-subunits, each comprised of six membrane-spanning domains (S1–S6) with a pore region and a voltage sensor. These tetramers are either homotetrameric (composed of a single type of α-subunit) or heterotetrameric, which contributes to the unique biophysical properties of individual channels. Functional diversity of KV activities is further enhanced by association of accessory proteins with KV tetramers that can greatly impact channel ontogeny and function. The accessory β-subunits are members of the aldose reductase superfamily of NADPH oxidoreductases, providing a surface interaction with several proteins, including protein kinases and phosphatases. Like α-subunits, each β-subunit has splice variants, contributing to tissue heterogeneity [10].
Activity of the 4-aminopyridine-sensitive KV channels in PASMCs play a pivotal role in regulating resting membrane potential, and [Ca2+]cyt, as well as in triggering hypoxia-mediated membrane depolarisation and pulmonary vasoconstriction [11–13]. Several members of the KV channel superfamily, including KV1.2, KV1.5, KV2.1, KV3.1 and KV9.3, have been found to be expressed by PASMCs [14–16]. Among these, particular attention has been given to KV1.5 (encoded by the KCNA5 gene), preferentially expressed in the small resistance pulmonary arteries rather than in conduit pulmonary arteries and diminished in PASMC following hypoxia exposure [14, 15]. Reduced expression of KV1.5 is a common denominator of human and experimental PAH suggesting an important role of this channel in the pathogenesis of various forms of PH [16–19]. Accordingly, in vivo gene transfer of KV1.5 was shown to reduce PH and to restore hypoxic pulmonary vasoconstriction in chronically hypoxic rats, adding further weight to its implication in PAH [20]. Although KV1.5 is considered as a potential therapeutic target, the molecular mechanisms leading to its reduced expression in this disease are not clear. Multiple mechanisms have been shown to contribute to its regulation (fig. 2).

{kind=link}
{kind=link}
Proposed schematic representation of pathways involved in the regulation of K+ current in pulmonary arterial hypertension pulmonary artery smooth muscle cells. [Ca2+]cyt: cytoplasmic free calcium concentration; Em: membrane potential; PI3K: phosphoinositide 3-kinase; PIP2: phosphatidylinositol-4,5-biphosphate; IP3: inositol-1,4,5-triphosphate; DAG: diacylglycerol; PLC: phospholipase C; PKC: protein kinase C; SrcTK: Src family tyrosine kinase; MLCK: myosin light-chain kinase; ROS: reactive oxygen species; HIF-1α: hypoxia-inducible factor-1α; PDK1: pyruvate dehydrogenase kinase, isozyme 1; HXK2: hexokinase 2; NFAT: nuclear factor of activated T-cells; STAT3: signal transducer and activator of transcription 3; SMAD: Mothers against decapentaplegic homologue; MAPK: mitogen-activated protein kinases; ψm: mitochondrial membrane potential; ET-R: endothelin receptor; BMP: bone morphogenetic protein; BMPRII: BMP receptor type II; TP: thromboxane receptor; 5-HTR: 5-hydroxytryptamine receptor; TNF-α: tumour necrosis factor-α; IL-6: interleukin-6; PDGF: platelet-derived growth factor; FGF: fibroblast growth factor; TNFR: TNF receptor; IL-6R: IL-6 receptor; RTK: receptor tyrosine kinases.
Activation of the phospholipase C–protein kinase C cascade regulates KV1.5 channel function
As mentioned earlier, pathogenesis of PAH is characterised by increased levels of pro-proliferative factors (serotonin, endothelin-1 and thromboxane A2 (TXA2)) and reduced levels of anti-proliferative factors, such as nitric oxide and prostacyclin, in the pulmonary vascular wall [2]. Decreased KV1.5-mediated current in PASMCs results from both direct effects of hypoxia and indirectly from hypoxia-induced expression of mitogenic factors.
The effects of serotonin (5-HT) on the pulmonary circulation have been of major interest since reports pinpointing the use of serotoninergic appetite suppressant drugs as a risk factor for the development of PAH [21]. The implication that serotonin is a key determinant of vessel remodelling was further confirmed by several observations highlighting: 1) high platelet and plasma levels of serotonin in patients with PAH [22], 2) prevention of PAH in animal models following inhibition of serotonin receptors [23, 24], and 3) a congenital predisposition to develop PAH in fawn-hooded rats caused by a defect in platelet serotonin storage [25]. Briefly, under pathological conditions, pulmonary endothelial cells produce high levels of serotonin that acts in a paracrine fashion on adjacent PASMCs to promote cell proliferation and contraction [26–28]. Results showing that exposure of human normotensive PASMCs to aminorex, fenfluramine or dexfenfluramide (appetite suppressant drugs that interact with the metabolism of serotonin) reduces the expression of KV1.5 and inhibits K+ current support the notion that serotonin signalling causes PAH, at least in part, through its effect on KV channel gene regulation [29, 30]. Cogolludo et al. [31] reported that serotonin treatment of PASMCs or Ltk− cells expressing cloned hKV1.5 markedly inhibited KV current and depolarised the cell membrane. Antagonists of 5-HT2A receptors abolished these effects. Under pathological circumstances, the binding of serotonin to the 5-HT2A receptors activates phospholipase C (PLC) through Gq family G-proteins that, in turn, lead to the cleavage of phosphatidylinositol-4,5-biphosphate (PIP2) into inositol-1,4,5-triphosphate and diacylglycerol (DAG) (fig. 2) [32, 33]. The former triggers the release of Ca2+ from intracellular pools and the latter activates protein kinase C (PKC), which phosphorylates several intracellular substrates [33]. Likewise, inhibitors of PLC, conventional PKC and tyrosine kinase prevented the effects of serotonin on KV currents [31]. The authors found that KV1.5 coimmunoprecipitated with 5-HT2A receptors and caveolin-1 in native pulmonary arteries underscoring that this physical interaction could serve to cluster the 5-HT2A receptor and signalling proteins with the K+ channel, allowing for specific modulation [31]. More importantly, Cogolludo et al. [31], demonstrated that stimulation with serotonin leads to caveolin-dependent internalisation of KV1.5, ensuring dynamic cell surface channel expression [31, 34]. Likewise, endocytosis of KV2.1 and KV1.5 channels has been reported to be a key mechanism in the control of KV channel activity, which is regulated by tyrosine phosphorylation mediated by Src kinase [35–37]. Thus, serotonin signalling appears to modulate KV current in PASMCs through direct alterations in KV1.5 trafficking and surface expression. This constitutes a mechanistic insight into how the levels of KV1.5 channels are downregulated in PAH (fig. 2).
TXA2 and prostacyclin are major arachidonic acid metabolites produced by vascular cells. TXA2 is a potent vasoconstrictor, whereas prostacyclin is a powerful vasodilator with antiplatelet and anti-proliferative effects. In both the primary and secondary forms of PH, the balance between these two molecules is shifted toward TXA2 [38]. Interestingly, a TXA2 analogue (U46619), through activation of the thromboxane endoperoxide receptor, inhibited KV current and depolarised PASMCs. Using a PKCζ pseudosubstrate inhibitory peptide, Cogolludo et al. [39] provided evidence for the role of this kinase as a link between thromboxane endoperoxide receptor activation and KV channel inhibition. The requirement for PKCζ in mediating TXA2-induced effects on KV current was further confirmed by the same group using PKCζ−/− pulmonary arteries [40]. In addition, sequestosome1/p62, a stress-inducible protein regulated by the redox-sensitive transcription factor Nrf2, was shown to serve as a scaffold protein acting as a physical link in the assembly of PKCζ–sequestosome1/p62–potassium channel complexes and to facilitate phosphorylation of KVβ by PKCζ, which induces inhibition of pulmonary arterial KV1.5 channels [41]. Consistent with this and similar to findings in PKCζ−/− PASMCs, U46619 fails to inhibit KV currents in PASMCs deficient for sequestosome1/p62 [40]. Collectively, these data provide evidence for a modulatory role of sequestosome1/p62 in the functional activity of KV1.5. It is worth noting that the subtypes of PKC involved in the regulation of PASMC KV1.5 channels vary among vasoconstrictors, since serotonin inhibits PASMC surface expression of KV1.5 channels in a PKCζ-independent manner, contrary to inhibition byTXA2 [31].
The interaction between growth factor receptors and the KV1.5 channel was examined in Xenopus oocytes, an advantageous model for studying connections between receptors and ion channels. Coexpression of cloned mouse platelet-derived growth factor (PDGF) receptor and cloned KV1.5 in Xenopus oocytes, followed by PDGF stimulation led to a decline in the KV1.5 current amplitude. Taking advantage of the fact that the fibroblast growth factor (FGF) and PDGF receptors share structural similarity and are thought to have a similar mechanism of activation, the involvement of PLCγ was explored using a mutated form of the FGF receptor that does not associate with PLCγ [42]. In coexpression experiments, the mutant FGF receptor was not able to modulate KV1.5 current amplitude, providing strong evidence that increased PLCγ activity is required for the reduction in KV1.5 current amplitude [42]. Considering the implication of FGF2 and PDGF in pulmonary vascular remodelling, the same mechanisms could conceivably occur in PAH PASMCs [43, 44].
Bone morphogenetic protein signalling and potassium channels
Several studies underscoring a critical role for bone morphogenetic protein (BMP) signalling in PAH have been published. A wide range of mutations, which are distributed throughout the coding region of the BMPR2 gene, have been identified in patients with PAH [45, 46]. Consistently, transgenic mice expressing a dominant-negative Bmpr2 gene in smooth muscle cells (SM22-tet-BMPR2delx4+) develop increased pulmonary arterial pressure, associated with a modest increase in arterial muscularisation [47]. To test whether anomalies seen in SM22-tet-BMPR2delx4+ are mediated by reduced KV channel expression, Young et al. [48] investigated the expression levels of several KV channels and revealed a pronounced diminution of KV1.5 protein expression in the lungs of transgenic mice. Consistent with this, treatment of PASMCs with recombinant BMP2 stimulates KV1.5 expression and was accompanied by increased current density in these cells; a functional effect blocked by an anti-KV1.5 antibody [48, 49]. The mechanism of KV1.5 regulation by BMP signalling is unclear. Considering that BMP2 induces apoptosis in human PASMCs by reducing the expression of the anti-apoptotic protein Bcl-2 and that overexpression of Bcl-2 diminishes KV1.5 expression, it is, therefore, conceivable that the functional effects of signalling on KV1.5 can be attributed to alteration of Bcl-2 [8, 50].
Transcriptional control: focus on hypoxia-inducible factor-1α and nuclear factor of activated T-cells
Although generation of reactive oxygen species (ROS) in mitochondria is known to be altered in PH, the direction of these changes remains debated [51]. A cancer-like metabolic shift towards a glycolytic metabolism even in the presence of adequate oxygen, also designated the Warburg effect, has been demonstrated as a pathogenic element that can contribute to the development of PAH [52]. Mitochondrial dysfunction, characterised by hyperpolarisation of mitochondrial membrane potential and fragmentation, generates a low ROS environment causing a pseudo-hypoxic state that rapidly stabilises and triggers nuclear translocation of the master transcription regulator of the adaptive response to hypoxia, hypoxia-inducible factor (HIF)-1α [18, 53]. The relationship between HIF-1α activation and KV1.5 expression was investigated in PAH patients and in fawn-hooded rats, which develop spontaneous PAH with ageing [18]. Normoxic HIF-1α activation (e.g. nuclear localisation) was almost universal in both cultured fawn-hooded rat PASMCs and human small pulmonary arteries from PAH patients. This correlates with a significant diminution of KV1.5 and K+ current, membrane depolarisation and increased cytosolic calcium. Furthermore, expression of KV1.5 and K+ current were restored in fawn-hooded rat PASMCs exposed to hyperoxia or transfected with an adenovirus coding for a dominant-negative form of HIF-1α, strengthening the fact that HIF-1α activation accounts for the downregulation of KV1.5 [18].
A sustained increase in cytosolic Ca2+, due to potassium-dependent or independent mechanisms, is an important activator of the nuclear factor of activated T-cells (NFAT) family of transcription factors. In resting cells, NFAT proteins are phosphorylated and reside in the cytoplasm. Upon stimulation, NFAT are dephosphorylated by calcineurin, leading to its translocation into the nucleus where it can regulate gene expression [54] (fig. 2). Persistent activation of NFATc2 is a hallmark of human idiopathic PAH (IPAH) PASMCs [55]. Inhibition of its nuclear translocation by VIVIT or cyclosporine A was shown to reverse KV1.5 downregulation in normal PASMCs exposed to hypoxia. Moreover, in vivo NFAT inhibition by cyclosporine A treatment was shown to restore KV1.5 expression and reverse right ventricular and pulmonary artery medial hypertrophy in the monocrotaline (MCT)-induced rat model of PAH [55]. In agreement with the direct involvement of NFATc2 in KV1.5 regulation, serotonin treatment of PASMCs induced marked nuclear translocation of NFATc2 and a significant decrease in KV1.5 protein expression [19]. Although analyses of the KCNA5 gene revealed multiple putative NFAT binding elements in the 5′ untranslated region, there is no evidence of direct transcriptional regulation.
Several other elements also contribute to transcriptional control of KCNA5. Among these, c-jun, a member of the transcription factor activator protein-1 family was demonstrated to influence voltage-gated K+ channel activity in PASMCs. By infecting PASMCs with an adenovirus expressing c-Jun, Yu et al. [56] demonstrated that overexpression of this transcription factor was associated with a significant decrease in the amplitude of KV current by downregulating KV1.5 expression.
Inflammation and potassium channels in PAH: an under studied field
Pulmonary vascular inflammation (chemokine-driven leukocyte accumulation in the pulmonary vascular lesions of PAH) and systemic inflammation (rise in blood levels of pro-inflammatory cytokines/chemokines, C-reactive protein and auto-antibodies) are a common feature of human and experimental PAH [57, 58]. Although inflammation is recognised to play a significant role in the pathogenesis of PAH, relative few studies have investigated the functional effects of pro-inflammatory cytokines on K+ current in PASMCs. Sutendra et al. [59] demonstrated that normal human PASMCs exposed to tumour necrosis factor (TNF)-α adopt a PAH phenotype with decreased K+ current. The molecular mechanisms mediating this effect remain unclear, but can be attributed to the activation of STAT3 (signal transducer and activator of transcription 3); a transcription factor accounting for the modulation of the expression of several proteins such as HIF-1α and NFAT (fig. 2) [60]. Indeed, increased STAT3 activation has been described in normal PASMCs treated with TNF-α or interleukin-6, suggesting that the decrease in K+ current elicited by these inflammatory factors is due, at least in part, to a STAT3-dependent pathway [61, 62].
Evidence for single nucleotide polymorphisms in the KCNA5 gene that underlie susceptibility to PAH
The identification of sequence polymorphisms is a key to understanding human genetic differences and diseases. The possible impacts of single nucleotide polymorphisms in KCNA5 on channel expression and function in PAH have been investigated [63, 64]. In taking advantage of large cohorts restricted to the European Caucasian population, a KCNA5 rs10744676 variant, located in the putative promoter of KCNA5, was found to be associated with systemic sclerosis (SSc). Interestingly, analysis within the SSc cohort revealed that a KCNA5 rs10744676 C allele was associated with protection from PAH [64]. It is suggested that this polymorphism within a CACCC box, normally recognised by transcription factors of the Sp/Krüppel-like factor family, could affect transcription factor binding and hence influence KCNA5 transcription and expression levels. The functional consequences of this polymorphism remain to be addressed. Nevertheless, combined deletion, site-directed mutagenesis, pharmacological and chromatin immunoprecipitation approaches have shown that the Kcna5 promoter was critically dependent on Sp1 regulation via CACCC box motifs in murine vascular smooth muscle cells, highlighting the functional importance of CACCC box motifs to drive KCNA5 gene expression [65].
Participation of microRNAs in the regulation of Kv1.5 in PASMCs
MicroRNAs are a class of short non-coding RNAs that act as negative regulators of gene expression by inhibiting the translation of target mRNAs, by promoting their degradation [66]. It is becoming increasingly apparent that the aberrant expression of microRNAs is causally related to a variety of disease states and PAH is no exception. Indeed, multiple deregulated microRNAs (e.g. miR17-92 cluster, miR-21, miR-143/145, miR-204 and miR-424/503) have been implicated in PAH [67, 68]. MicroRNA expression profiling between IPAH-PASMCs and normal PASMCs and subsequent bioinformatics microRNA target gene prediction were recently performed to examine the potential role of microRNAs in the downregulated KV channel expression and whole-cell KV current, characteristic of IPAH-PASMCs. Among microRNAs found to be selectively upregulated in IPAH-PASMCs, miR-29b was selected based on the predicted miR-29b–KV1.5 mRNA relationships. Forced expression of miR-29b in normal PASMCs decreased Kv1.5 protein expression, whereas inhibition of miR-29b in IPAH-PASMCs, using antisense oligonucleotides, rescued Kv1.5 protein levels, demonstrating that KV1.5 is a direct target of miR-29b in IPAH-PASMCs. These findings highlight an additional level of complexity to the regulation of KV1.5 (fig. 2) [69].
Post-translational modifications of KV1.5: implications in PAH?
Protein post-translational modification (PTM) is a highly dynamic and crucial mechanism in which the functional properties of a protein are altered by the covalent addition of a chemical group or protein to its amino-acid residues. Independently of PAH, KV1.5 has been shown to undergo diverse, reversible or irreversible PTM, including phosphorylation, S-acylation, palmitoylation, sumoylation, ubiquitination and glycosylation, which impact on ion channel expression, channel trafficking to the membrane, stability and activity [35, 70–74]. In response to oxidative stress, a sulfenic acid modification has been reported on a cysteine residue present in the C-terminal domain of KV1.5 that triggers internalisation of KV1.5, decreases current density, and diverts the channel from the recycling endosome towards degradation [75]. Moreover, tyrosine phosphorylation of KV1.5 and redox-sensitive KV1.5 sumoylation were also reported leading to its inactivation [35, 73]. Using Xenopus oocytes as a model to study ion channels in a controlled in vivo environment, it was demonstrated that expression of the ubiquitin ligase Nedd4-2 declined KV1.5 currents by ubiquitinating and thereby reducing KV1.5 plasma membrane expression [74]. Finally, transfection of a KV1.5 palmitoylation-deficient mutant into Chinese hamster ovary cells has been shown to enhance the expression of surface current through enhanced localisation of the channel to the plasma membrane, indicating that palmitoylation reduced KV1.5 biological function [71]. No information is available on the PTM status of KV1.5 channels in PAH. Taking into account that metabolic pathways provide substrates for PTM and that PAH is recognised as a disorder associated with significant metabolic dysfunction, it can be assumed that major alterations in PTM of K+ channels occur in PAH [76, 77]. Nonetheless, the likely importance of these regulatory mechanisms remains to be explored.
Together, these findings demonstrate that KV1.5 is carefully regulated throughout the lifecycle of the channel and that alterations of these regulatory mechanisms can have important functional repercussions on KV1.5-mediated currents and PAH progression.
Two-pore domain potassium channels in PAH
K2P channels are subdivided into six families, based on their sequence similarities and functional resemblance: TWIK, TREK, TASK, TALK, THIK, and TRESK [78]. Because the K2P channels are typically open at negative membrane potential, they have been strongly implicated in establishing the background K+ conductance known to stabilise the negative resting membrane potential and counterbalance depolarisation [78]. KCNK3 (also called TASK1) encodes a pH-sensitive K2P channel, which has been shown to be expressed by PASMCs and to contribute significantly to the resting membrane potential [79]. Indeed, TASK1 knockdown in human PASMCs caused a depolarisation of the resting membrane potential [80]. In direct connection with PAH, TASK1 was documented as being inhibited in human PASMCs by endothelin-1 through an endothelin receptor type A–PLC–PIP2–DAG–PKC-dependent signalling cascade [81]. In addition, hypoxia was shown to reduce the phosphorylation level of TASK1 and to inhibit TASK1 current in reducing the active phosphorylated state of the Src family tyrosine kinase (SrcTK). Inhibition of SrcTK depolarises PASMCs and reduces KV and KCa currents [82]. This finding supports the observation that dasatinib, a dual Src/Abl kinase inhibitor, can cause PAH [83]. Moreover, it is important to point out that the KCNK3 channel was fully inhibited by serotonin in neurons suggesting that this mechanism may operate in PASMCs [84]. In keeping with previous findings that decreased efflux of K+ from the cell is associated with resistance to apoptosis, overexpression of KCNK3 in neurons induces cell death, while its inactivation protects against cell death [85]. In addition to KCNK3, KCNK6 (also designated TWIK2), another member of the K2P family, has recently been shown to be involved in PH. Kcnk6-deficient mice present with an increase in right ventricular systolic pressure, indicating the development of vascular remodelling with PH [86]. Collectively, these findings provide compelling evidence that KCNK3 and KCNK6 work in concert with other oxygen-sensitive K+ channels to regulate pulmonary vascular tone.
KCNK3 (TASK1) as a cause of PAH
Familial cases of PAH have long been recognised and are usually due to mutations in members of the transforming growth factor (TGF) signalling cascade [45, 46, 87]. BMPR2 mutations account for ∼70% of familial PAH (HPAH) and 15% of patients with IPAH. To a lesser extent, heritable and idiopathic PAH are also associated with mutations of genes related to TGF-β signalling, such as activin receptor-like kinase 1 (ALK1), endoglin (ENG), and mothers against decapentaplegic 9 (SMAD9) [88]. To date, a significant proportion of familial cases of PAH have not been linked to a genetic cause. Recent advances in genome sequencing technologies have provided unprecedented opportunities to identify mutations. Whole exome sequencing has gained popularity and was recently and successfully applied in a family in which multiple members presented PAH without identifiable mutations in any of the genes known to be linked to the disease. This approach allowed the discovery of KCNK3 as a new predisposing gene for PAH [89]. In screening additional patients with IPAH and HPAH, a total of six KCNK3 variants were identified, all of which resulted in a loss of function of the channel at physiological pH that probably causes depolarisation of the resting membrane potential. The prevalence of KCNK3 mutations was 1.3% in IPAH and 3.2% in HPAH [89]. This study provides the first causal relationship between a K+ channel and PAH, and consequently PAH is now considered as a channelopathy. Surprisingly, neither right ventricular hypertrophy nor vessel remodelling were observed in Kcnk3−/− mice underlying the fact that pulmonary arterial pressure of mutant mice was not elevated [90]. Divergence between human and rodent orthologous genes, existence of functional redundancy in rodents, and inter-species variability in expression levels may explain the apparent differences. In particular, KCNK3 does not form a functional channel in mouse PASMCs [91]. In mice, Kcnk3 function is probably replaced by other K+ channels, for example Kcnk6. Accordingly, Kcnk6 mutant mice are more sensitive to hypoxia-induced PH [86]. Although involvement of KCNK3 is probable in human PAH, analysis of its expression level in animal models mimicking the human disease is required. The generation of a Kcnk3 mutant rat, a popular model species in the field of PH, will constitute a powerful tool to decipher its implication in vascular remodelling.
Potassium channels: therapeutic opportunities
Until recently, the treatment of PAH has focused on drugs that target vasoconstriction pathways. Since current vasodilator therapies are limited, research is now focusing on strategies that could reverse structural remodelling in the pulmonary arterial wall. Based on advances made in the physiopathogenesis of PAH, the anti-remodelling impact of multiple pharmacological agents have been tested in experimental models, and restoration of pulmonary artery K+ channel expression and K+ current are the parameters most often explored as a readout of their beneficial effects. For instance, dichloroacetate (DCA), which enhances oxidative phosphorylation by inhibiting the mitochondrial pyruvate dehydrogenase kinase, has been shown to prevent and reverse PAH in the MCT rat model. DCA treatment almost completely reversed the downregulation of KV1.5 observed in isolated PASMCs from MCT rats, improving K+ current [17]. Similar results were observed in the MCT model using cyclosporine-A (an inhibitor of calcineurin–NFATc binding), fluoxetine (a selective serotonin reuptake inhibitor), and Plumbagin (a STAT3 inhibitor) [55, 91, 92]. Moreover, treatment with a selective survivin inhibitor was recently reported to significantly reduce pulmonary arterial pressure and to increase KV1.5 transcript levels in rats exposed to chronic hypoxia [93]. Based on published data showing that, in both human and experimental animal models of PAH, KV1.5 is mainly subjected to transcriptional and intracellular protein trafficking regulations leading to reduced surface membrane expression, the clinical benefit of selective Kv channel openers appears questionable. As development of PAH is a multifactorial process, the disease should be approached from different therapeutic angles. This provides a strong rationale for combination therapy.
Conclusion
Numerous factors involved in PAH pathogenesis have been shown to impact K+ channels at multiple levels. Reduced K+ channel activity in PASMCs promotes cell proliferation, resistance to apoptosis and vasoconstriction contributing to vascular remodelling. Although the precise mechanisms by which KCNK3 leads to PAH remain largely unknown, its importance in controlling vascular functions offers great promise for future research. Molecules promoting indirect KCNA5 and KCNK3 expression, or direct gene transfer may constitute valuable therapeutic strategies for the treatment of PAH.
Footnotes
Conflict of interest: None declared.
- Received May 20, 2015.
- Accepted July 9, 2015.
- Copyright ©ERS 2015
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Overview of potassium channels
- Potassium channels in regulation of cell proliferation and cell death
- KV channel dysfunction as a contributing factor in the development of PAH
- Two-pore domain potassium channels in PAH
- Potassium channels: therapeutic opportunities
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics