





Chronic thromboembolic pulmonary hypertension (CTEPH) is a rare but notoriously underdiagnosed complication of pulmonary embolism, which carries a poor prognosis if left untreated. CTEPH results from the obstruction of the pulmonary vascular bed by fibro-thrombotic material, which may completely occlude the lumen. Although massive or recurrent pulmonary embolism is thought to be the initiating event, a significant proportion of patients have no history of symptomatic pulmonary embolism and only a few patients with acute pulmonary embolism develop CTEPH [1]. As a consequence, the true prevalence and incidence of CTEPH remain unknown [2]. Current data, retrieved from registries, suggest that the incidence of CTEPH averages between three and 30 per million in the general population [3], and that the proportion of patients developing CTEPH after acute pulmonary embolism may vary between 0.1% and 9.1% [4]. The standard and potentially curative treatment for CTEPH is a surgical procedure, known as pulmonary endarterectomy (PEA) [5], which involves removing organised thrombi, neointima and media inner layers obstructing the pulmonary arteries (fig. 1b) [6]. PEA, when performed in experienced centres and on selected patients, shows low perioperative mortality, and provides major improvements in haemodynamics, symptoms and survival [7]. Moreover, CTEPH is a “dual” pulmonary vascular disorder combining major vessel obstruction and small vessel arteriopathy, which is histologically indistinguishable from idiopathic pulmonary arterial hypertension (PAH) in the non-occluded lung areas [8]. As a consequence, PAH-targeted therapy, including prostacyclin analogues, endothelin-1 receptor antagonists and phosphodiesterase-5 inhibitors, has been largely used in CTEPH [9] and should be considered for inoperable patients or patients with persistent pulmonary hypertension after PEA [10]. More recently, riociguat, a stimulator of soluble guanylate cyclase, has been approved for the treatment of inoperable CTEPH and persistent pulmonary hypertension after PEA, on the basis of demonstrated improvements in haemodynamics and exercise capacity [11].

{kind=link}
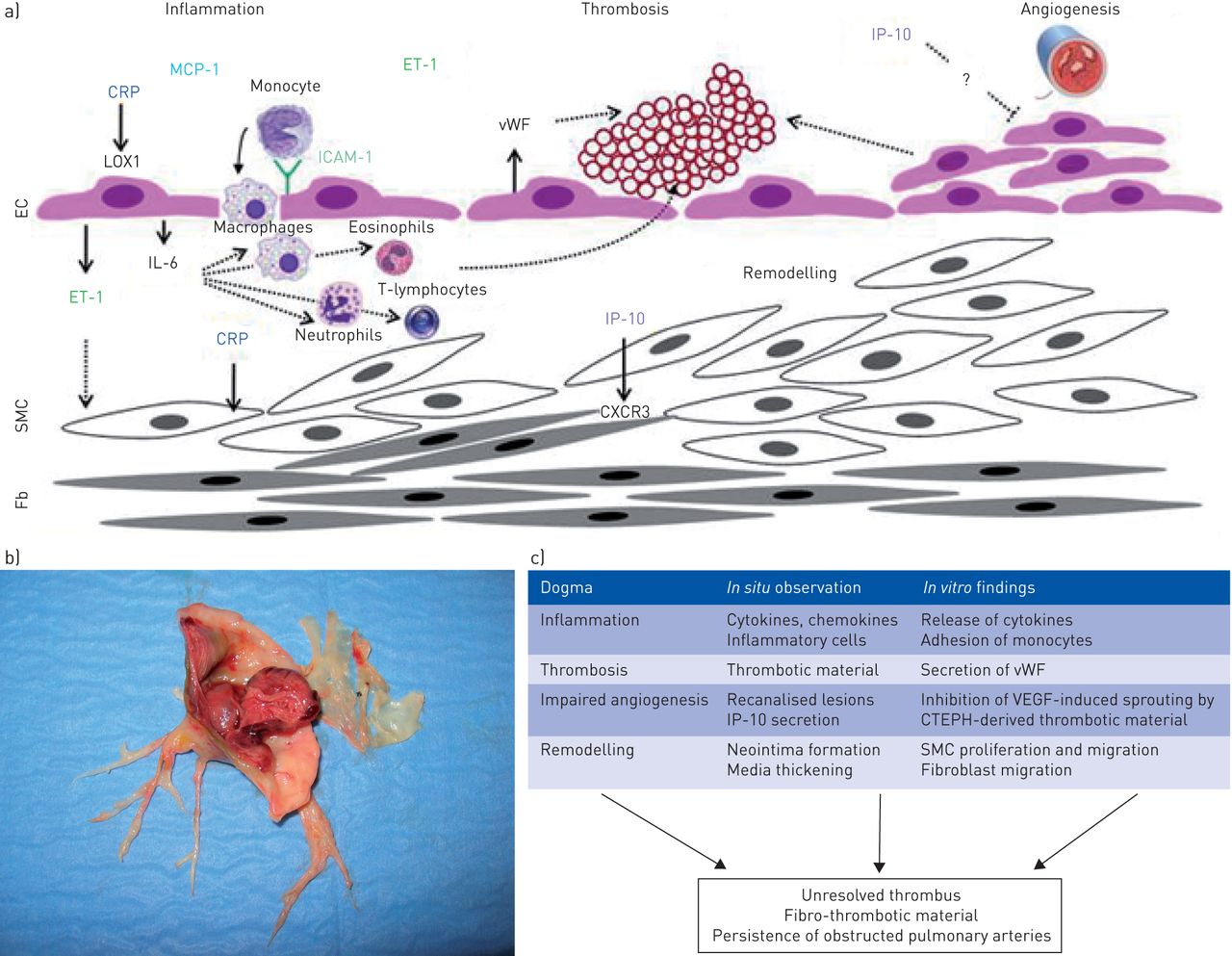
a) Schematic view of potential mechanisms contributing to persistent vessel obstruction and chronic thromboembolic pulmonary hypertension (CTEPH) pathogenesis. b) Fibro-thrombotic material obstructing pulmonary arteries collected during pulmonary endarterectomy. c) Information from in situ to in vitro findings. MCP-1: monocyte chemoattractant protein; CRP: C-reactive protein; LOX1: lectin-type oxidised low-density lipoprotein receptor; ET-1: endothelin-1; IL-6: interleukin-6; ICAM-1: intercellular adhesion molecule-1; SMC: smooth muscle cell; Fb: fibroblasts; vWF: von Willebrand Factor; IP-10: interferon-γ-induced protein-10; VEGF: vascular endothelial growth factor.
The reasons why acute embolic obstruction does not resolve and causes pulmonary hypertension in a minority of patients remain unclear. Incomplete resolution of thrombi is often observed after acute pulmonary embolism [12] and patients displaying persistent occlusions also have higher mean pulmonary arterial pressure compared with patients without any defect [13]. However, pressures usually remain within the normal range and patients do not show any exercise limitation. Various medical conditions, such as splenectomy, chronic inflammatory disorders, ventriculoatrial shunts, infections or cancer, are associated with CTEPH [4], and CTEPH is more common in people with non-O blood groups [14, 15]. In addition to an impaired balance between coagulation and fibrinolysis, and deficient angiogenesis with impaired thrombus recanalisation, inflammatory thrombosis has been suggested to be involved in the pathogenesis of CTEPH [3] (fig. 1a). Although the role of inflammation in the pathogenesis of PAH has been extensively documented both in PAH animal models and patients, the involvement of the inflammatory process in the pathogenesis of CTEPH remains elusive.
In this issue of the European Respiratory Journal, Zabini et al. [16] hypothesise that cytokines could mediate the obstructive remodelling of pulmonary arteries in CTEPH. They profiled a broad range of cytokines, including interleukin (IL)-8, IL-6, interferon-γ-induced protein (IP)-10, monocyte chemoattractant protein (MCP)-1, monokine induced by interferon-γ (MIG), RANTES, CX3CL1, macrophage inflammatory protein (MIP)1α and CXCL12, in PEA tissue, PEA tissue supernatant and in serum of newly diagnosed CTEPH patients, and identified IP-10 as a potential pathogenic factor for CTEPH. IP-10, also known as CXCL10, is a small cytokine belonging to the CXC chemokine family that is secreted in response to interferon (IFN)-γ by several cell types, including monocytes, endothelial cells and fibroblasts [17]. IP-10 is a chemoattractant for monocytes/macrophages, T-cells, natural killer cells and dendritic cells. It promotes T-cell adhesion to endothelial cells and inhibits bone marrow colony formation and angiogenesis [18, 19].
The authors used a translational approach combining analysis of PEA tissue, serum from CTEPH patients and in vitro assays with clinical parameters to show that IP-10 is negatively correlated with the 6-min walking distance (6MWD), the primary end-point used in most randomised clinical trials, and with cardiac output. The authors also found a correlation between circulating IL-6 and right atrial pressure (RAP), pulmonary vascular resistance (PVR) and N-terminal pro-brain natriuretic peptide (NT-proBNP). Kimura et al. [20] previously showed a correlation between plasma and tissue expression of the CCL2 chemokine MCP-1 with elevated PVR. Elevated circulating C-reactive protein (CRP) levels, observed in unselected CTEPH patients, were significantly decreased following PEA [21]. Similarly, in this latest CTEPH cohort (n=127), plasma CRP levels significantly correlated with RAP and negatively with 6MWD (unpublished data). In the aforementioned studies, inflammatory cytokines could be markers of right heart function failure rather than vascular disease.
Interestingly, the authors suggested a pathogenic role for IP-10 by showing in vitro inhibition of IP-10-induced fibroblast migration by antibodies against CXCR3, the receptor for IP-10. A bundle of converging evidence further supports the hypothesis that IP-10 and IFN-γ signalling pathways are potential key players in the pathogenesis of CTEPH (fig. 1a) and not simply innocent bystanders: 1) medical conditions with an inflammatory component, including staphylococcal infection, splenectomy, inflammatory bowel disease and osteomyelitis, are risk factors for CTEPH [22], and are associated with IP-10 secretion and/or elevated circulating IP-10 [23–25]; 2) IFN-γ signalling could determine the relative pathogenicity of Staphylococcus aureus strains [26]; 3) venous thrombus resolution is misguided in the presence of S. aureus infection [27]; 4) IP-10 is an inhibitor of angiogenesis; and 5) defective angiogenesis delaying thrombus resolution is potentially involved in CTEPH [28], and could be promoted by splenectomy [29]. In our hands, CRP also seemed to contribute to the disease process by activating the nuclear factor (NF)-κB pathway, thereby inducing endothelial cell dysfunction in CTEPH (fig. 1a) [30, 31]. The inflammatory hypothesis is further supported by the presence of eosinophils, macrophages, neutrophils and T-lymphocytes in the vascular material removed during surgery, as shown by Zabini et al. [16] and by previous studies (fig. 1a and c) [6, 32, 33].
The exploratory study by Zabini et al. [16] was conducted on a limited number of patients, although clinically well-characterised and consistently sex- and age-matched with healthy controls. The authors are aware of this limitation and propose further investigations extended to a larger number of patients in a prospective design. Accordingly, analysis of different potential inflammatory candidate mediators and development of therapeutic strategies targeting inflammation in this rare disease would require united efforts, possibly leaning on registries like the CTEPH European registry [1] and its worldwide extension (www.cteph-association.org/registries/cteph-registry). The authors remain very cautious regarding the impact of their study on possible therapeutic strategies, limiting themselves to speculating on the potential role of inflammatory mediators, such as IP-10, in the mechanisms underlying progressive obstruction of the pulmonary arteries in CTEPH (fig. 1a). However, the study by Zabini et al. [16] displays other limitations: patient tissues and plasma samples were from separate cohorts, the origin of the supernatants was not clearly specified, and one may also wonder whether the healthy matched subjects were an optimal control population and whether idiopathic PAH patients would have been a better control cohort. In addition, it is unlikely to be possible to rule out any inflammatory disease and/or malignancy in a 60-year-old cohort.
Furthermore, most of the aforementioned studies have been performed using blood and tissue samples from patients at advanced stages of the disease. Prospective long-term follow-up studies gathering large numbers of acute pulmonary embolism patients, and combining clinical, biological and environmental parameters, are necessary to validate these pathogenic hypotheses. Similarly, development of accurate and relevant animal models clearly mimicking CTEPH pathobiology is needed, in addition to the recently developed models of abnormal venous thrombus resolution [28, 29] or of large vessel mechanical obstruction [34, 35].
Even if we do believe that inflammation is a potential therapeutic target in CTEPH, we are not yet ready for prime time. Actually, IP-10 is one potential player among many others and evidence that blocking the IP-10 receptor could alter disease progression and prevent or reverse advanced obstructive lesions (fig. 1b) is still missing.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received June 30, 2014.
- Accepted July 23, 2014.
- ©ERS 2014
References










