





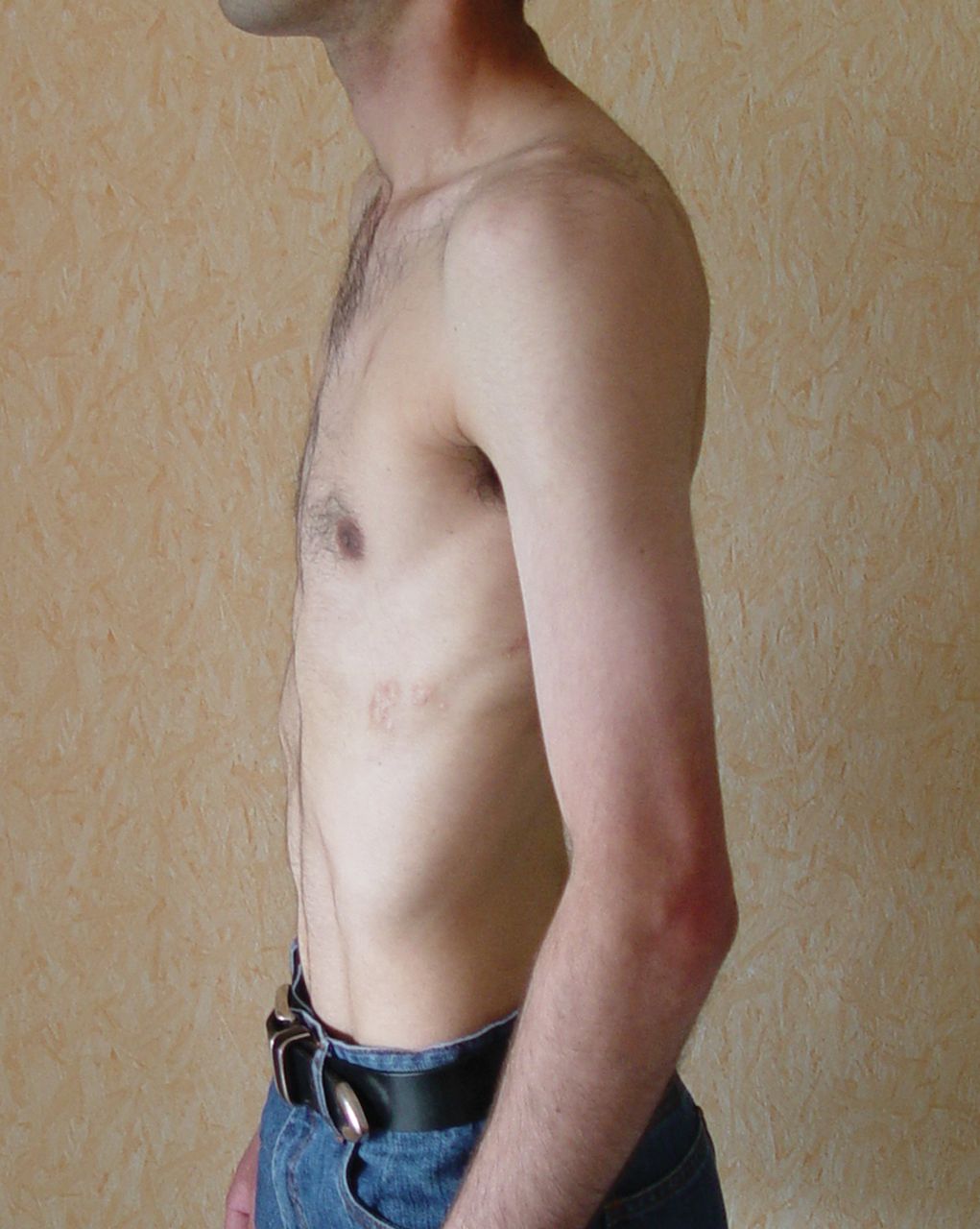
In this issue of the European Respiratory Journal, Beynat-Mouterde et al. [1] report on six young adults (three of whom were female) who developed a clinical imaging pattern of predominant upper lobe fibrosis with apical pneumothoraces (fig. 1). Presentation in all six patients was similar with cough, dyspnoea, occasional chest pain and weight loss. Imaging was distinctive and showed a cephalad, irregular, pleural-based thickening that encroached on the lung bilaterally. Five patients presented with “platythorax” (fig. 2), a preferential reduction in the anterio-posterior diameter of the chest wall. In all patients, severe restrictive or restrictive-obstructive lung dysfunction progressed to hypoxaemic and hypercarbic respiratory failure, which was fatal in four patients despite intensive care unit support. This outcome was all the more tragic as these four patients had survived previous malignant conditions, including haematological malignancy (n=3) and brain tumour (n=1).
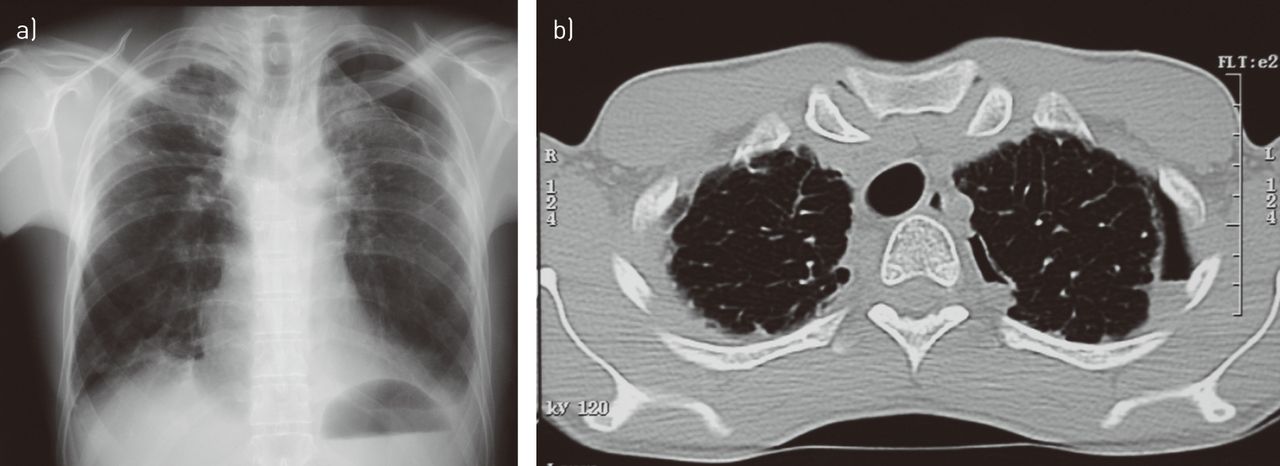
a) Frontal chest radiograph in a patient following chemotherapy for acute lymphoblastic leukaemia showing a typical appearance of pleuropulmonary fibroelastosis (corresponding to case 2 in [1]). Note the markedly thickened pleural dome on the right and partial spontaneous pneumothorax on the left. b) Computed tomography scan of the same patient taken several months later. A chest tube had been inserted and removed in the interim. A smaller, persistent pneumothorax is visible.

Advanced cases may show markedly restrictive physiology and reduced anterio-posterior diameter of the thorax; a trait known as platythorax.
Pathology in four patients showed a pattern consistent with pleuropulmonary fibroelastosis (PPFE). Due to similar clinical imaging presentation, two additional patients without biopsy were included in the study of Beynat-Mouterde et al. [1]. There was a history (6 months to 16 years previously) of multi-agent chemotherapy in all six patients; the alkylators cyclophosphamide (n=5) and BCNU (bis-chloroethylnitrosourea; n=1) were the common denominators in their treatment. Beynat-Mouterde et al. [1] raised the possibility that alkylating drugs may have triggered or caused PPFE. Issues raised by their study relate to: 1) PPFE as a clinically, radiographically and pathologically separate and recognisable entity; 2) possible aetiological factors at the origin of PPFE, including drugs; and 3) current management strategies.
PPFE (as reviewed recently [2]) is an unusual interstitial lung disease with circumscribed fibrosis of the pleura and subjacent lung, which is of interest to pathologists [3–8], pulmonologists [3, 4, 6–8] and radiologists [3–6, 8, 9]. Although elastosis has also been described in the heart and skin, it appeared to be confined clinically and on imaging to the pleura and lung in cases of PPFE. The condition does not clearly fit into any of the previously described interstitial pneumonias and, thus, is now specifically included in the classification of idiopathic interstitial pneumonias (IIP) under the heading “rare IIP” [2, 10]. The six patients in the study by Beynat-Mouterde et al. [1] are reminiscent of the 78 cases (59 in the English literature) of PPFE that have been published since the initial reports from Japanese investigators (to whom credit is due), under the term “upper lobe fibrosis” or “Amitani’s disease” [11–13], as well as initial reports in the western world by Frankel et al. [3]. The three largest series of PPFE included nine [14] 12 [8] and 15 [15] cases, respectively. PPFE emerges as a distinctive array of clinical, imaging and pathological abnormalities affecting the apices and lateral aspects of the lung and pleura bilaterally. The condition has a variable prognosis with substantial mortality [2, 8]. Of potential teleological interest, a nearly identical condition has recently been described in a third of aged donkeys at necropsy [16, 17]. Of note, PPFE affects the dorsal aspect of both lungs in the donkey [17, 18], which is the nondependent part of the lung in quadrupeds. In humans PPFE affects the lung apices which are the nondependent parts of the human lung [18].
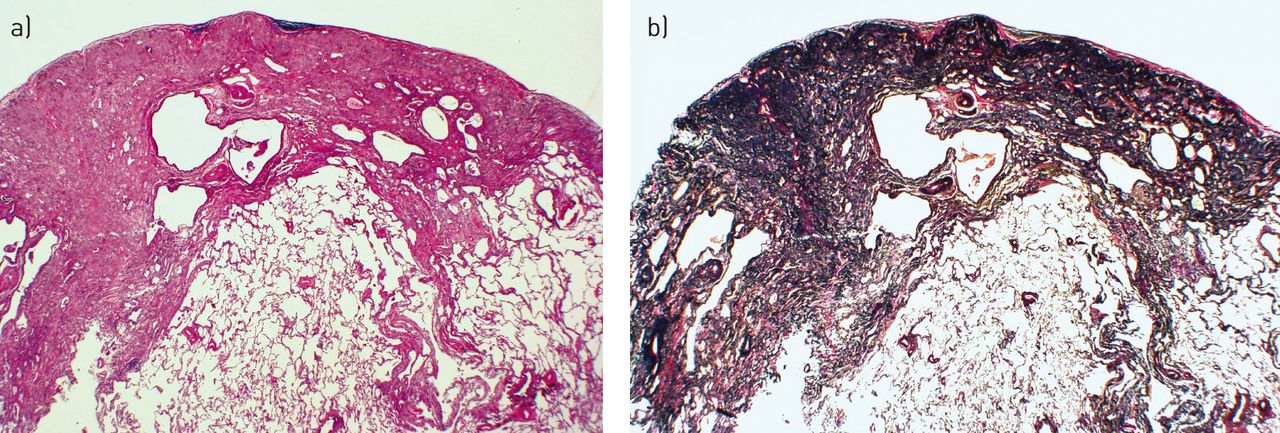
As the name implies, PPFE is a morphologically descriptive term denoting intense elastotic fibrosis of the visceral pleura and subjacent lung and collagenous fibrosis rich in haphazardly arranged elastic fibres (figs 3 and 4). Elastosis is a distinctive form of chronic scarring in the lung that differs from the common scarring seen in idiopathic pulmonary fibrosis (IPF). PPFE lungs contain twice as much elastin compared to IPF [19]. The process may extend into the septa and deeper into the lung (figs 3 and 4) [8, 20]. Elastic fibres are best visualised using orcein or Verhoeff van Gieson elastic stain (fig. 4b). The histopathological diagnosis of PPFE can still be considered without performing the specific latter elastic fibre stains, because the features on Haematoxylin and Eosin are characteristic once one is familiar with them. On microscopy, the boundary between the pleuropulmonary fibrotic area and the relatively spared juxtaposed lung is characteristically sharp [2–4, 6–8, 21]. Associated pathological features include an absence of classic honeycombing, inconspicuous fibroblastic foci, a sparse mononuclear lymphocytic infiltrate and moderate interstitial fibrosis in the remaining lung. Diffuse alveolar damage, alveolar haemorrhage [15] and/or obliterative bronchiolitis [6, 15] may be concomitant findings in patients who have had lung or bone marrow/haematopoietic stem cell transplantation [15, 22]. As a generalisation, both the pleural surface and the underlying lung are less affected or appear normal in the lower zones. The changes on gross pathology match those on imaging (figs 3 and 4) [8, 23, 24].

Gross pathology of the lung showing grey elastotic fibrosis in the subpleural regions and around the bronchovascular bundles.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histology staining showing a sharply demarcated zone of subpleural fibrosis with abundant elastic tissue on elastic tissue stain. a) Haematoxylin and Eosin stain and b) Verhoeff van Gieson elastic stain. Scanning power microscopy ∼40×.
Clinically, idiopathic PPFE affects males and females equally, and preferentially nonsmokers with a proportion of patients being relatively young [2]. Compared to IPF, PPFE patients have a lower body mass index and more severe restrictive lung dysfunction [19]. Onset in childhood is associated with poorer outcome [20, 25], possibly because the restricted fibrotic pleura and lung significantly impede lung growth [20, 25]. Presenting features include chronic dull pleuritic pain with episodes of a sharper quality (which may not reflect pneumothorax in all patients), platythorax [26] (which can be measured on computed tomography (CT) or with a calliper), and small chronic spontaneous uni- or bilateral pneumothoraces [27]. On imaging, the fibrotic pleura and subjacent collapsed and fibrotic lung manifest as a dense irregular peripheral rim [1, 8]. Wedge-shaped pleural-based densities protrude along parenchymal septa toward the hila [8, 9], and the latter tend to be retracted upwards (figs 1 and 3) [24]. Calcifications have not been reported within the thickened pleura [8], which helps to separate PPFE from asbestos-induced pleural thickening; a condition that may also affect the apical pleura, although very rarely [28]. In PPFE, there are usually features of established fibrosis away from the subpleural regions in the more central parts of the upper lobes on CT [8, 24]. Although it may take years for PPFE to develop before patients become symptomatic and seek medical attention [1], some cases develop and progress rapidly. Once PPFE becomes symptomatic, patients may remain stable for a long period of time or progress inexorably to hypercarbic respiratory failure. Then, outcome is poor despite ventilatory support with a 40–66% mortality rate in a few years with or without an identifiable abrupt exacerbation [1, 2, 6, 8, 25, 29]. Spontaneous partial, apical, uni- or bilateral pneumothorax is a marker of abnormality for PPFE, and is present at some point in ∼30% of overall patients [2, 30]. This complication was seen in five out of the six patients in the study by Beynat-Mouterde et al. [1]. Some pneumothoraces are small and may be simply observed [9]. Suggested mechanisms leading to pneumothoraces include cysts present in the apical fibrotic area, altered resistance of the pleura to shear stress, parenchymal bullae in the lung transplant recipient and air trapping upstream to obliterative bronchiolitis. Pneumothoraces rarely absorb spontaneously, and persistent air leak and poor re-expansion of the underlying lung are common [2]. Lung or pleural biopsy must be avoided. Many consider it unnecessary in cases that fit clinically and radiologically for PPFE. In addition, post-operative iatrogenic pneumothorax commonly complicates the procedure [1, 8]. To the extent that the biopsy will provide more than a mere academic diagnosis, it should be considered solely as a companion procedure if surgical cure of pneumothorax is contemplated. While a definitive diagnosis of PPFE traditionally requires histological examination of the pleuropulmonary interface, a sizeable number of the patients will not undergo the procedure. The reasons for refraining from doing so include: very advanced disease; poor ventilatory reserve; lack of perceived benefit as no treatment is available for the condition; and the fact that the disease can be strongly suspected on clinical and radiological grounds. For those cases where no biopsy is available, a label of “consistent with PPFE” has been proposed [2, 8], with the acronym PPFE being reserved for pathologically documented cases [8]. A study in donkeys has shown the potential merit of chest ultrasonography as a noninvasive test to diagnose PPFE (table 1) [17]. Since the disease has now been well defined, it is likely that more and more patients will be diagnosed with PPFE noninvasively.
However suggestive the clinical and imaging presentation of PPFE is, other aetiologies of pleural thickening should be considered [31, 32], including: tuberculosis and tuberculosis pneumothorax treatment; aspergillosis [1, 33], although aspergillosis may complicate the course of established PPFE [33] as shown in one patient in [1]; other infections; connective tissue disease with rheumatoid arthritis lupus and ankylosing spondylitis [31]; ulcerative colitis [26]; haemothorax; a history of coronary artery bypass graft [34]; exposure to asbestos [35]; and rare or orphan pleural disease [32]. In most of these conditions, pleural thickening has a predilection to involve the lung bases or to predominate on one side, as opposed to PPFE. The differential diagnosis is usually resolved by: reviewing earlier imaging; searching for extrathoracic involvement that is typically absent in PPFE; appropriate laboratory tests for tuberculosis, aspergillosis, other infections and connective tissue diseases; and mineral dust study of bronchoalveolar lavage fluid. Asbestos pleural thickening is typically more prominent in the parietal pleura and shows the histological characteristic basket-weave collagenous appearance without any admixture of elastotic and fibrotic tissue [36].
The apical cap is a well-known, generally idiopathic lesion of the lung apices that is preferentially found in males of older age [37–39]. The apical cap is in the form of a subpleural pyramidal or spiculated scar spanning 0.7 to 5.2 cm in diameter. Apical caps can be large and simulate a neoplasm [37], but they do not involve the pleura circumferentially like PPFE. On pathological investigation, the cap also corresponds to dense collagen and curls or folded elastic fibres [37–39]. Time may tell whether the apical cap and PPFE share more than morphologic similarity.
Regarding aetiology, the majority of PPFE cases (>50%) have been reported as a late complication of bone marrow/haematopoietic stem cell [2, 6] and lung transplantation [2, 6, 40], for which the terms “upper lobe fibrosis” [29, 40] and “chronic lung allograft dysfunction” [41] have been applied. The prevalence of PPFE in lung transplant recipients may be as high as 2% [29]. Rare familial occurrences of “consistent with” and bona fide PPFE have been described in young individuals, mostly females [3, 42]. 10% of PPFE cases developed in patients previously exposed to chemotherapeutic agents, sometimes many years after management of the primary disease [1, 2, 20, 25, 43].
Drugs are a cause célèbre of respiratory injury [44]. Pleural reactions have been known to be a result of drug injury since the 1960s [45, 46], accounting for 7% of all respiratory reactions to drugs [44]. Pleural reactions include an effusion with or without pleural eosinophilia, antinuclear-antibody positive effusion (the lupus syndrome), haemothorax, acute pleuritis and pleural thickening [44]. Reactions to ergots (e.g. bromocritptine, ergotamine, lisuride, and methysergide) may result in pleural effusion and/or pleural and pericardial thickening or fibrosis [44, 46, 47]. Individuals with a history of exposure to asbestos may be at a higher risk of developing pleural thickening if exposed to ergot drugs [48, 49]. However, ergot-induced pleural disease differs from PPFE in that pleural thickening usually localises at the lung bases and develops during therapy with the agent; although the pleura rarely returns to a normal state, pleural disease does improve after drug discontinuation [47, 48]. Furthermore, there is no evidence for increased elastic fibres on pathology in ergot-induced pleural thickening [47].
Cases of PPFE and cases which appear to be consistent with PPFE as a result of chronic complications of BCNU treatment [20, 23, 25, 50–52] or cyclophosphamide [3, 43, 53, 54] used to treat breast cancer [3], brain tumours [20, 23, 51] or granulomatous polyangiitis [53] have been previously published, supporting the findings of Beynat-Mouterde et al. [1]. Studies by O’Driscoll and co-workers [20, 25], Taylor et al. [23] and Hasleton et al. [51] have detailed the clinical, imaging and pathological features of late BCNU toxicity. The disease develops after a symptom-free interval of 2–12 years. Affected individuals present with the characteristic clinical, imaging and pathological features of PPFE, including pneumothorax and interstitial elastosis [20, 51]. Craniospinal irradiation in those patients with a brain tumour was not thought to contribute to their disease as the dose received to the chest was minimal [20]. Similarly, Malik et al. [43] reported on five cases of late pleuropulmonary toxicity developing 6 months to 6 years after termination of treatment with cyclophosphamide. Apical pleuropulmonary fibrosis involvement was also observed. Three out of the five patients died due to respiratory failure [43]. The authors reviewed five similar cases from the literature [43]. All the previously reported BCNU and cyclophosphamide cases closely resemble those reported by Beynat-Mouterde et al. [1].
Although appealing, the nature of an association of alkylating agents and PPFE, either causal or circumstantial, needs be examined carefully [55]. The case against causal association, includes the possibility that PPFE can develop idiopathically or, as some reports suggest, as an abreaction to infectious agents [2], as well as: due to the lack of a clear dose–response curve; development and progression of fibrosis after a free interval of several years after termination of treatment; lack of reversal with drug withdrawal; and absence of robust epidemiological support or absence of a suitable animal model [55]. Current models of pleural fibrosis use transforming growth factor-β1, bleomycin and carbon black instilled in the pleural space as the inciting agents [56, 57]. Conversely, in favour of an association between alkylating agents and PPFE are: the normalcy of pre-therapy chest imaging in the study by Beynat-Mouterde et al. [1]; the homogenous and consistent reports of the same distinctive drug-associated disease from different countries over the past 35 years [2]; the notion that two separate alkylating agents can produce an association with PPFE; and the occurrence of PPFE in young individuals [20, 25] in whom fibrotic conditions are relatively rare. Evidence for drug-associated elastosis in organs other than the pleura and lung is extremely scarce, with only one reported case of endocardial fibroelastosis following therapy with adriamycin [58].
Management of this dreadful condition is difficult. Corticosteroids and immunosuppressants offer transitory or no improvement, even less cure; except in a very few cases. Pleuropulmonary biopsy should be undertaken very cautiously, if at all. Experience with decortication is extremely limited [59]. Timely consultation with a transplant team is warranted, as transplantation has been suggested [4, 5] and successfully attempted in a few cases [42, 60].
To conclude, the imaging features of PPFE are very suggestive, if not pathognomonic. PPFE is a probable if not frequent late complication of chemotherapy regimens containing alkylating agents. As greater awareness of PPFE will lead to more frequent and hopefully more accurate diagnosis and reporting, we can only hope this will improve our comprehension of PPFE and its causes, including drug therapy, enabling novel treatment approaches to be identified, thus, improving the currently poor outcome.
Acknowledgments
We would like to thank Nicolas Baudouin (Dept of Pulmonary Medicine and Intensive Care, Centre Hospitalier Universitaire Le Bocage, Dijon, France), who diligently cared for several patients in our intensive care unit who were included in the study by C. Beynat-Mouterde and co-workers. Title in tribute to Lou Reed (1942–2013).
Footnotes
Conflict of interest: None declared.
- Received May 14, 2014.
- Accepted July 1, 2014.
- ©ERS 2014
References










