





Abstract
Fluticasone furoate/vilanterol trifenatate (FF/VI) is a once-daily inhaled corticosteroid/long-acting β2-agonist combination in development for chronic obstructive pulmonary disease (COPD) treatment. We compared the efficacy and safety of FF/VI versus fluticasone propionate/salmeterol (FP/SAL) twice daily over 12 weeks.
Moderate to very severe COPD patients received FF/VI 100/25 μg once daily in the morning (n=266) or FP/SAL 500/50 μg twice daily (n=262). The primary end-point was a change from baseline in 0–24 h weighted mean forced expiratory volume in 1 s (wmFEV1) at 12 weeks. Additional end-points included time to 100 mL improvement from baseline on day 1 and a change from baseline in St George's Respiratory Questionnaire (SGRQ). Safety was also assessed.
wmFEV1 (mean 130 mL) was greater and time to 100 mL improvement shorter (median 16 min) with FF/VI than FP/SAL (weighted mean 108 mL, median 28 min). Health status (SGRQ total score) improved in both groups (FF/VI -4.3 units, FP/SAL -3.0 units). Differences between treatments were not statistically significant. Six patients in the FF/VI (2%) and three in the FP/SAL (1%) arm experienced serious adverse events, none of which were considered to be drug related.
Improvements in lung function and health status were not significantly different between FF/VI 100/25 μg once daily and FP/SAL 500/50 μg twice daily; there was no apparent difference between the safety profiles of either therapy.
Abstract
Once-daily fluticasone furoate/vilanterol trifenatate and twice-daily fluticasone propionate/salmeterol act similarly http://ow.ly/sdj0x
Introduction
In patients with moderate to very severe chronic obstructive pulmonary disease (COPD), combined treatment with an inhaled corticosteroid (ICS) and a long-acting β2 agonist (LABA) improves lung function [1] and health status [2], reduces the annual exacerbation rate [3] and slows disease progression as assessed by the rate of decline of forced expiratory volume in 1 s (FEV1) [4]. Currently available ICS/LABA combinations require twice-daily dosing (e.g. fluticasone propionate (FP) and salmeterol (SAL)).
Fluticasone furoate (FF)/vilanterol trifenatate (VI) is a novel ICS/LABA combination being developed for once-daily administration, a dosing scheme that may improve adherence [5]. This is potentially important, as low therapeutic compliance is associated with poor clinical outcomes in COPD [6, 7]. FF is structurally distinct from FP and has enhanced glucocorticoid receptor binding [8]. In vitro, VI exhibits ≥1000-fold selectivity for the β2 receptor over the β1 or β3 receptors [9]. Furthermore VI is an ante-drug [10], which degrades to metabolites with reduced/negligible potency upon entry to the systemic circulation. In vitro, the main metabolites of VI are ≥2500-fold less potent than VI at the β2 receptor [11].
Phase IIb dose-ranging trials of VI in COPD [12] and FF in asthma [13–15] have shown that both therapies are efficacious and exhibit a once-daily dosing profile [16, 17]. FF/VI has been studied at strengths of 50/25 μg, 100/25 μg and 200/25 μg in a small 4-week crossover study [18], and in larger 24-week lung function studies [19, 20] and 52-week exacerbation studies [21]; none of these studies compared FF/VI with an established ICS/LABA combination, such as FP/SAL. The current study is the first to compare head-to-head the efficacy and safety of FF/VI (100/25 μg once daily) versus FP/SAL (500/50 μg twice daily), conducted over 12 weeks of treatment in patients with moderate to very severe COPD.
Methods
Patients and ethics
We studied male and female adults aged ≥40 years, with a smoking history of ≥10 pack-years and a post-bronchodilator (salbutamol) FEV1/forced vital capacity ratio of ≤0.70 and a FEV1 ≤70% predicted [22]. Patients had to have experienced at least one moderate COPD exacerbation (requiring treatment with oral corticosteroid/antibiotic) or severe exacerbation (leading to hospitalisation) within the past 3 years. Patients with a current diagnosis of asthma, serious underlying disease or infections, hospitalisation due to COPD within 12 weeks of screening, or acute worsening of COPD (defined as use of corticosteroids or antibiotics) within 6 weeks of screening were not included. All patients signed their informed consent, and the protocol was approved by local ethics review committees of the 61 centres where the study was conducted. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Study design
This was a 12-week, randomised, multicentre (61 centres in Europe and Asia), double-blind, double-dummy, parallel-group, comparative efficacy/safety study. The study is registered at www.clinicaltrials.gov (identifier number NCT01342913); GlaxoSmithKline study number HZC113107.
Following screening, patients entered a 2-week, placebo run-in period, after which they were randomised in a 1:1 ratio to receive in a double-blind manner FF/VI 100/25 μg once daily in the morning via the ELLIPTA dry powder inhaler (GlaxoSmithKline, Ware, UK), which emits a dose of 92 μg FF and 22 μg VI, or FP/SAL 500/50 μg twice daily via the Accuhaler (GlaxoSmithKline, Uxbridge, UK), for 12 weeks. Patients receiving FF/VI also took a placebo via Accuhaler once in the morning and once in the evening, and patients receiving FP/SAL took a placebo once in the morning using the ELLIPTA dry powder inhaler. Patient randomisation schedule was computer generated by GlaxoSmithKline, and treatments were assigned via a telephone call to the interactive voice response system using RandAll and RAMOS (both GlaxoSmithKline, London, UK). To account for a potential confounding effect of bronchial reversibility, randomisation was stratified based upon the patient's reversibility, defined as a change in FEV1 of ≥12% and ≥200 mL 10–15 min after four inhalations of salbutamol. Reversibility was assessed at the screening visit, prior to the 2-week placebo run-in period. For standardisation across all subjects, study-supplied salbutamol was used and administered via a study-supplied valved holding chamber (AeroChamber Plus; GlaxoSmithKline) for the purpose of reversibility testing. Compliance with treatment was assessed by reviewing the dose counters on both inhalers at randomisation (day 1), day 28, day 56 and on day 84. Salbutamol was supplied to patients for symptomatic relief during the study. Ipratropium, mucolytics and oxygen for ≤12 h per day were permitted provided their dose did not change during the study. Likewise, cardioselective β-blockers, intranasal sodium cromoglycate or nedocromil sodium, intranasal corticosteroids, diuretics and medications for other disorders were also permitted, provided the dose remained constant wherever possible and their use was not expected to affect lung function. Medications not permitted during the course of the study are provided in the online supplementary material.
Efficacy assessment
The primary efficacy end-point of the study was the 24-h effect of FF/VI on lung function after 12 weeks of treatment (day 84), as compared with FP/SAL. This was assessed through the change from baseline in weighted mean (wm) FEV1. Baseline FEV1 was defined as the mean of two FEV1 assessments taken 30 min and 5 min pre-dose on day 1 of the study (i.e. after the 2-week placebo run-in period). Day 84 FEV1 was defined as the weighted mean of 15 FEV1 assessments taken at 5, 15, 30 and 60 min pre-dose, and at 2, 4, 6, 8, 12, 13, 14, 16, 20 and 24 h post-dose. All FEV1 assessments were conducted using standardised equipment that met or exceeded the suggested standards [23]. All sites were issued with Biomedical Systems Vitalograph 6800 Fleisch pneumotach (Biomedical Systems (BMS), Brussels, Belgium) for spirometry assessments prior to study start, and received training from BMS. ECG data was also obtained from BMS programming and equipment. Each site electronically transmitted both the spirometry and ECG data to BMS, where the data underwent quality control and best test review by the over-readers who were certified by BMS to analyse both the pulmonary function and ECG data.
Secondary efficacy end-points were: 1) time to 100 mL increase from baseline from 0–4 h on day 1; and 2) change from baseline in trough FEV1 on day 85, i.e. the comparison of the FEV1 recorded 24 h post-dose on day 84 with the baseline measure.
Other efficacy end-points included changes in health status, as determined by St George's Respiratory Questionnaire (SGRQ) for COPD and rescue-free 24 h periods. Health status was assessed on day 1 (i.e. following the 2-week placebo run-in) and day 84 of the study, or at the point of early withdrawal if prior to day 84. Finally, a post hoc analysis of the difference in lung function 0–4 h, 0–12 h and 12–24 h post-dose on day 84 was also conducted.
Safety assessment
Safety and tolerability were assessed by the incidence of adverse events and severe adverse events, coded using the Medical Dictionary for Regulatory Activities. Adverse events of special interest were also assessed. These comprised adverse events known to be associated with ICS and/or LABA therapy and included bone disorders, cardiovascular effects, effects on potassium, effects on glucose, hypersensitivity, local steroid effects, ocular effects, pneumonia, lower respiratory tract infections excluding pneumonia, systemic steroid effects and tremors. The incidence of COPD exacerbations and pneumonias, as well as abnormalities of oropharyngeal examinations, clinical chemistry and haematology assessments, vital signs and ECG measurements, were also recorded.
Statistical analysis
We assumed a treatment difference (FF/VI–FP/SAL) of 60 mL for 24 h wmFEV1 with a standard deviation of 190 mL. The 60 mL treatment difference in wmFEV1 on which the study was powered was based on extrapolation of previous wmFEV1 results with SAL [1] and VI [12] taking into account the putative minimal clinically important difference for trough FEV1 [24, 25]. Based upon this analysis, a 60 mL treatment difference in wmFEV1 was determined to be an appropriate difference for superiority of one active treatment compared to another (for further explanation please see online supplementary material). Accordingly, we calculated that a sample size of 212 evaluable patients in each arm of the study was needed to detect a statistically significant difference at a 5% level (two-sided) with 90% power. Assuming a withdrawal rate of 15%, we aimed to randomise a minimum of 250 patients per treatment arm.
Data are shown as mean±sd or proportion, as appropriate. The two treatment arms were compared using ANCOVA, with efficacy results displayed as mean±sd. To account for multiplicity across primary and secondary efficacy end-points, a p-value <0.05 was required for the primary end-point to allow statistical significance to be inferred for secondary end-points. For this reason a p-value is presented only for the primary comparison; otherwise, point estimates and 95% confidence intervals are presented, but no formal statistical comparison can be done. All efficacy and safety measurements were analysed in the intent-to-treat (ITT) population, as defined by all patients who were randomised to treatment and who received at least one dose of study medication.
Results
Patient characterisation
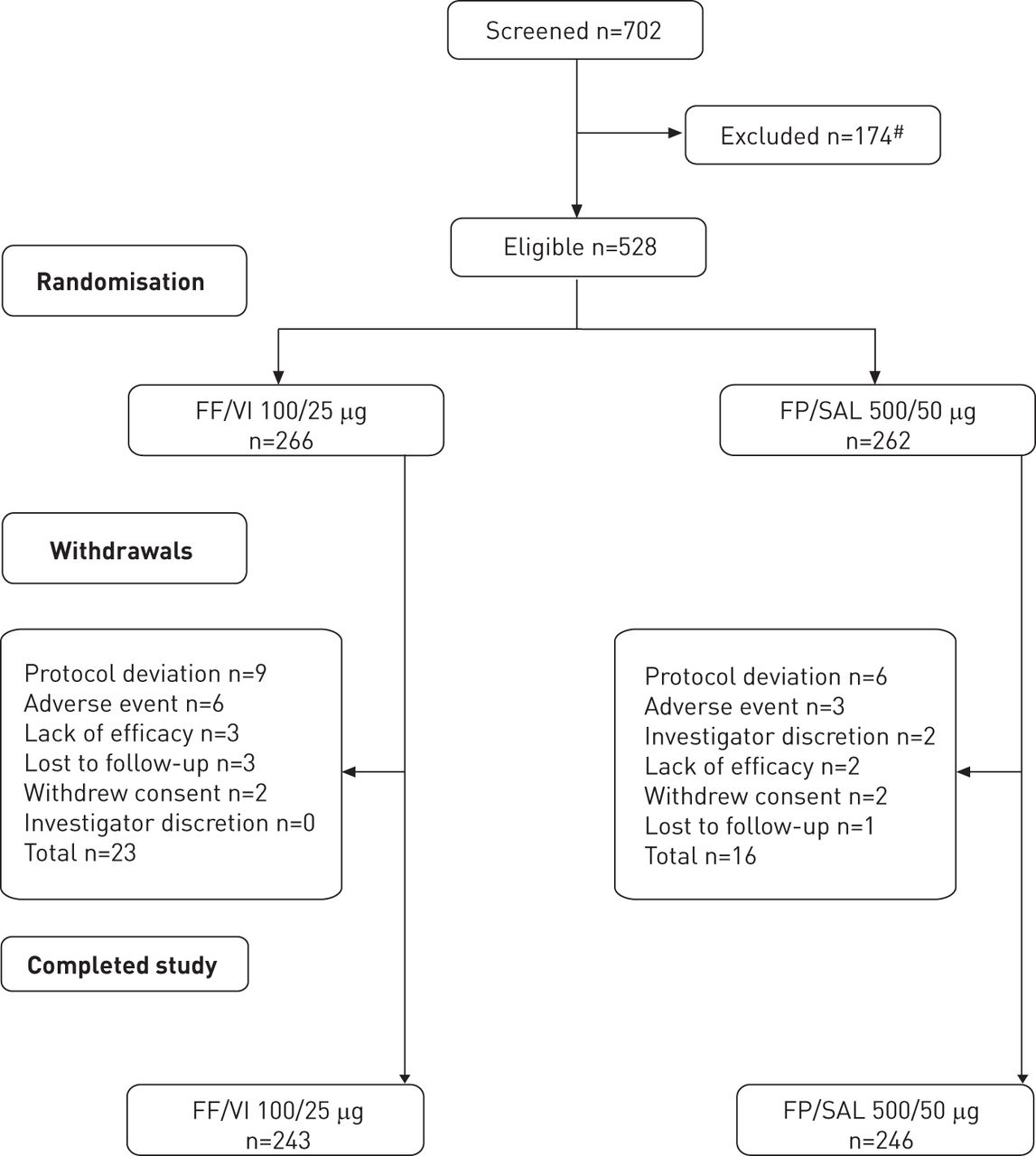
Figure 1 presents the consort diagram of the study. A total of 528 patients were randomised and comprised the ITT population. 243 (91%) patients in the FF/VI arm and 246 (94%) in the FP/SAL arm completed the study. The demographic and clinical characteristics of participants (ITT population), including age, sex, ethnicity, body mass index, comorbidities, medications received and mean FEV1 were similar at baseline regardless of study arm (table 1).
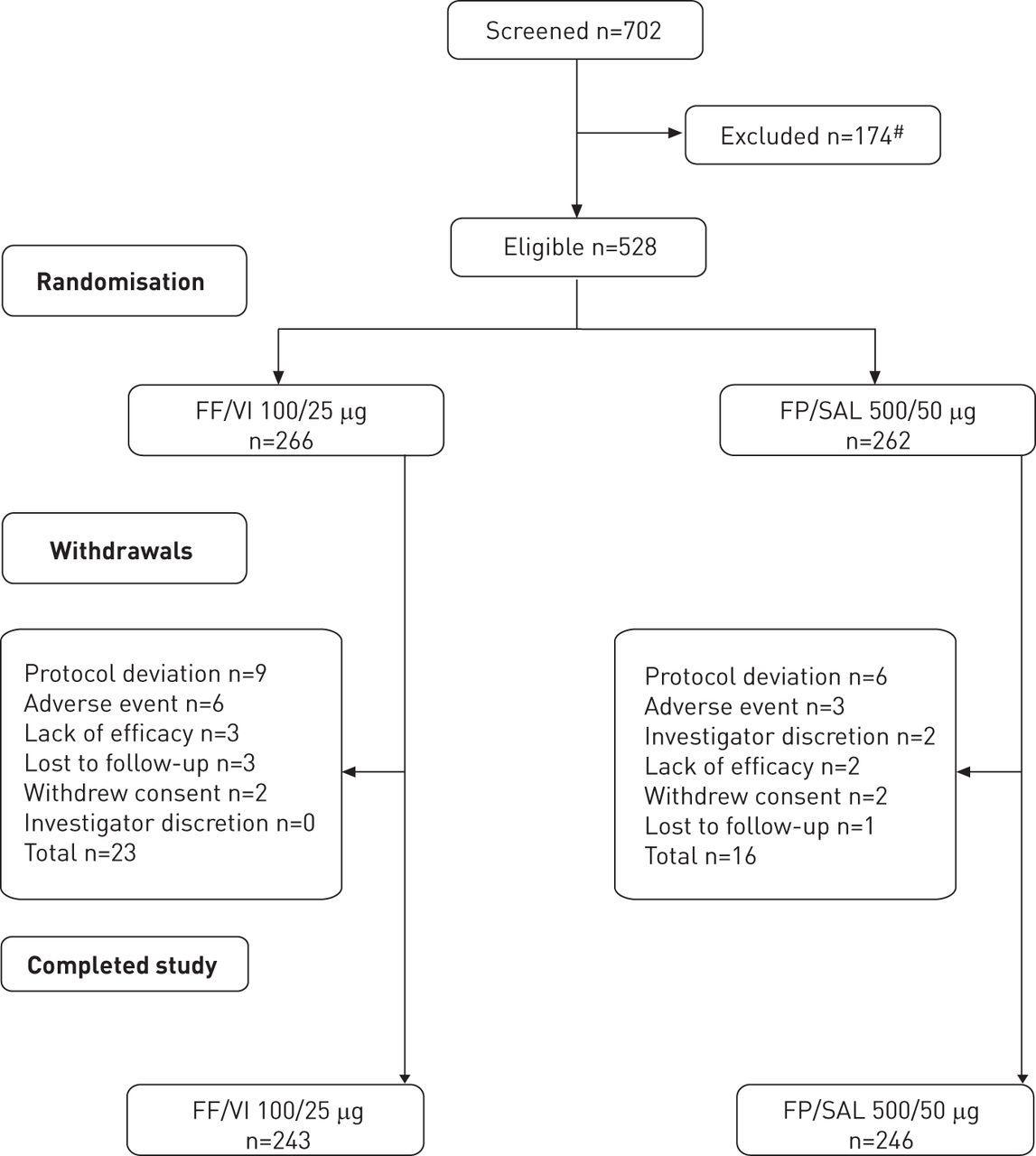
Consort diagram of the study. FF: fluticasone furoate; FP: fluticasone propionate; SAL: salmeterol; VI: vilanterol trifenatate. #: reasons for exclusion comprised: did not meet inclusion/met exclusion criteria (n=66), did not meet continuation criteria (n=89), withdrew consent (n=7), investgator discretion (n=6), protocol deviation (n=5) and adverse event (n=2). Subjects could be excluded from randomisation for more than one reason.
Efficacy: primary end-point
An improvement from baseline in 0–24 h wmFEV1 on day 84 was observed with both FF/VI (mean±sd 130±222 mL) and FP/SAL (108±221 mL); the difference in improvement between the two arms (22 mL) did not reach statistical significance (p=0.282). A post hoc comparison of changes in wmFEV1 between arms did show a difference from 0–4 h and 0–12 h post-dose (daytime), but not from 12–24 h post-dose (table 2 and fig. 2).

{kind=link}
{kind=link}
Change from baseline in the least squares mean forced expiratory volume in 1 s (FEV1) on day 84 in the intent-to-treat population. FF: fluticasone furoate; VI: vilanterol trifenatate; FP: fluticasone propionate; SAL: salmeterol.
Efficacy: secondary end-points
As explained in the methods section, because statistical significance was not achieved for the primary end-point, the analysis plan established that statistical significance could not be inferred for comparisons of secondary efficacy end-points. Hence, all differences described below should be considered as descriptive only.
The mean change from baseline in trough FEV1 on day 85 (an indicator of 24-h effect) was 111 mL in the FF/VI and 88 mL in the FP/SAL arms (ITT population) with a mean treatment difference of 23 mL (95% CI -20–66) (table 2). The median time to reach an increase in FEV1 of ≥100 mL on day 1 was 16 min in the FF/VI treatment group and 28 min in the FP/SAL treatment group. The minimal clinically important difference [26] for change from baseline in SGRQ was achieved in the FF/VI group (mean±sd -4.3±11.8) but not in the FP/SAL group (-3.0±11.8) (table 2). The proportion of rescue-free 24-h periods was similar between treatments (61.6% for FF/VI and 58.5% for FP/SAL during the first week of the study, and 62.5% for FF/VI and 59.8% for FP/SAL over the entire duration of the study).
Safety
The occurrence of on-treatment adverse events and drug-related adverse events was similar between the two study arms (table 3). More adverse events leading to withdrawal and more on-treatment severe adverse events were observed with FF/VI than with FP/SAL (table 3). Headache and nasopharyngitis were the most frequently reported on-treatment adverse events. The most frequently reported drug-related adverse event was oral candidiasis (preferred term) (FF/VI, n=2 and FP/SAL, n=4). Two out of the three adverse events leading to withdrawal in the FP/SAL arm were due to pneumonia (versus one event in the FF/VI arm), and three out of the six adverse events leading to withdrawal in the FF/VI arm were due to cardiac disorders (versus none in the FP/SAL arm), specifically atrial fibrillation (n=2) and congestive heart failure (n=1). None of the adverse events leading to withdrawal and none of the on-treatment severe adverse events were considered by the study investigators to be treatment related. There were no fatal on-treatment adverse events; one fatal event during the post-treatment follow-up period of FF/VI treatment (congestive heart failure) was not considered to be treatment related.
The occurrence of adverse events of special interest was generally low and similar across the two treatment groups, although more patients experienced cardiovascular adverse events in the FF/VI group (nine versus one) and more patients experienced local steroid effects in the FP/SAL group (10 versus three). Of the local steroid effects, three events each of oral candidiasis and oropharyngeal candidiasis, and one event of dysphonia were considered to be drug-related in the FP/SAL group, while two events of oral candidiasis and one event of dysphonia were considered to be drug-related in the FF/VI group (table 3). COPD exacerbations occurred in six (2%) patients receiving FF/VI and seven (3%) patients receiving FP/SAL. All but two of these patients were withdrawn from the study and all exacerbations resolved satisfactorily.
Three patients experienced four pneumonia events during the study (one from the FF/VI group and two from the FP/SAL group); one subject in the FP/SAL group experienced two pneumonias prior to being withdrawn. The protocol required that patients be withdrawn in the event of pneumonia; this subject should have been withdrawn at their first pneumonia but was not and so was considered a protocol violator. No abnormalities in laboratory values of potential clinical concern were observed during this study. There was no significant difference in pulse rate between treatment groups, either at randomisation or after 12 weeks of treatment (data not shown).
Discussion
This study shows that the efficacy and safety of the novel once-daily combination of FF/VI 100/25 μg in patients with moderate to very severe COPD over 12 weeks is not significantly different to that of the currently available FP/SAL 500/50 μg twice daily dose.
An ICS/LABA combination is a well-established therapeutic strategy for patients with COPD [27]. The improvements from baseline in lung function over 12 weeks with FF/VI and FP/SAL both exceeded 100 mL, which is regarded as a clinically perceptible difference [24, 25], although the 22 mL difference between the two treatment arms was not statistically significant. However, two observations (fig. 2) deserve comment. First, the FEV1 profile in the early part of the day appeared enhanced with FF/VI. This observation merits confirmation in other studies, as it is possible that it could translate into additional clinical benefits, such as occurrence of COPD exacerbations [28]. Secondly, the expected second peak in the FEV1 area under the curve observed after the administration of FP/SAL (fig. 2) did not surpass that of FF/VI, probably reflecting the pharmacological potency of FF/VI.
As recently emphasised by the Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease 2011 document, the assessment and treatment of symptoms is an important component of COPD management [27]. The SGRQ for COPD is a well-established instrument for the assessment of health status in patients with COPD [29]. A difference of ≥4 points in the SGRQ total score versus placebo at study end, or ≥4 points from baseline is considered to be the minimal clinically important difference for this measure [26]. We observed that treatment with FF/VI over 12 weeks demonstrated an improvement of ≥4 units (mean change 4.3), while FP/SAL achieved an improvement of <4 units (mean change 3.0). This supports a positive effect of FF/VI on patient's health status, but as the comparison is the change from baseline as opposed to a difference from placebo in change from baseline (there was no placebo arm in the current study), the results must be interpreted in light of this. Indeed, a placebo effect on health status has previously been noted in other interventional clinical trials in COPD [30, 31], possibly as a consequence of the overall health benefits of the regular clinic visits associated with participation in a clinical trial.
To further compare the 24-h efficacy profile of the two treatment arms, we performed a post hoc analysis of wmFEV1 at 0–4 h, 0–12 h and 12–24 h post-dose on day 84. We observed a larger wmFEV1 with FF/VI than with FP/SAL in both early periods, i.e. during the daytime. This may theoretically contribute to improved exercise tolerance and health-status, but requires further prospective assessment. During the second 12 h period on day 84 there was a 1-mL difference in wmFEV1 between FF/VI (88 mL) and FP/SAL (87 mL). It is important to recall that the 88 mL change from baseline recorded with FF/VI during this period occurred without the need for a second dose of therapy. As with any putative daytime effects, the potential impact of lung function changes upon night-time symptoms in COPD is unclear [32] and requires additional prospective assessment.
FF/VI differs from other currently available ICS/LABA combinations in that it is taken once daily whereas others require twice-daily dosing [33]. This simplified dosing regimen may improve adherence [34, 35], and consequently has the potential to provide better disease control and improved outcomes [36]. However, the present study was not designed to assess adherence to therapy and any relationship it may have to outcome. Indeed, mean adherence in both study arms, as assessed by evaluation of inhaler dose counters, was 97.5%, which suggests that the results observed herein reflect those achieved with optimal adherence. Whether such adherence would be observed in a real-world setting, where adherence to COPD therapy is known to be much lower [37], and whether dosing frequency would impact adherence and potentially outcome, remains to be established, ideally in the setting of an effectiveness study.
The rates of adverse events and severe adverse events were similar in both study arms; however, there were differences between the study arms in terms of adverse events of specific interest. 1) More local effects of steroids were observed in the FP/SAL arm than in the FF/VI arm. This may potentially be due to lower daily ICS exposure with FF/VI (100 μg·day−1) than with FP/SAL (1000 μg·day−1), although it must be considered that budesonide equivalent values for FF have not yet been determined (in asthma the daily budesonide dose equivalent to 1000 μg of FP is 1600 μg) [38]. 2) More cardiovascular effects were observed in the FF/VI arm than in the FP/SAL arm. Overall, LABA-mediated effects, such as those on potassium and glucose, or tremors, occurred infrequently with either treatment, and there was little difference in their incidence between the study arms. In addition, two 6-month trials with FF/VI 100/25 μg did not show an increase in cardiovascular adverse events compared with placebo [19, 20].
Our study did have some limitations. The absence of a placebo arm limited the interpretation of data for all end-points. Also, the relatively short duration of 12 weeks limited our ability to draw firm conclusions with respect to exacerbations, pneumonia and other systemic adverse events, as longer follow-up is normally required for differences in the incidence of these events to become apparent. Furthermore, there was no attempt to document potential pneumonia events radiologically. It is possible, therefore, that a larger sample size and/or an unselected COPD population may have identified any rare adverse events that were not observed in our relatively small and restricted population.
In conclusion, FF/VI (100/25 μg once daily) and FP/SAL (500/50 μg twice daily) both produced improvements from baseline in lung function and health status, in patients with moderate to very severe COPD; there was no statistical difference between the improvements observed with FP/SAL and those observed with FF/VI. The safety profile of each treatment did not markedly differ in terms of adverse events and severe adverse events, though more local steroid effects were observed with FP/SAL and more cardiovascular effects were observed with FF/VI. These findings suggest that FF/VI 100/25 μg provides an improvement in lung function and health status that is not superior to, but is comparable to, that provided by FP/SAL 500/50 μg, and furthermore is provided by once-daily as opposed to twice-daily therapy.
Acknowledgments
The authors thank participants for their willingness to contribute to medical research and field investigators for their dedication and quality of their work.
Editorial support in the form of development of an outline and first draft under the guidance of the lead author, copy-editing of drafts of this manuscript and generation of tables and figures was conducted by Geoff Weller (Gardiner-Caldwell Communications, Macclesfield, UK).
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Clinical trial: This study is registered at www.clinicaltrials.gov with identifier number NCT01342913; GlaxoSmithKline Study Code HZC113107.
Support statement: This work was funded by GlaxoSmithKline. Editorial support from G. Weller (Gardiner-Caldwell Communications, Macclesfield, UK) was also funded by GlaxoSmithKline.
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
- Received March 26, 2013.
- Accepted July 30, 2013.
- ©ERS 2014
References










