





Abstract
Idiopathic pulmonary fibrosis (IPF) is an ageing-related lung disorder characterised by expansion of the myofibroblast population and aberrant lung remodelling. Dehydroepiandrosterone (DHEA), a steroid pro-hormone, decreases with age but an exaggerated decline has been associated with chronic degenerative diseases.
We quantified the plasma levels of DHEA and its sulfated form (DHEA-S) in 137 IPF patients and 58 controls and examined the effects of DHEA on human lung fibroblasts.
Plasma DHEA/DHEA-S was significantly decreased in male IPF patients (median (range) DHEA: 4.4 (0.2–29.2) versus 6.7 (2.1–15.2) ng·mL−1, p<0.01; DHEA-S: 47 (15.0–211) versus 85.2 (37.6–247.0) μg·dL−1, p<0.001), while in females only DHEA-S was significantly decreased (32.6 (15.0–303.0) versus 68.3 (16.4–171) μg·dL−1, p<0.001). DHEA caused a decrease in fibroblast proliferation and an approximately two-fold increase in fibroblast apoptosis, probably through the intrinsic pathway with activation of caspase-9. This effect was accompanied by upregulation of several pro-apoptotic proteins (Bax and cyclin-dependent kinase-inhibitor CDNK1A) and downregulation of anti-apoptotic proteins, such as cellular inhibitor of apoptosis (c-IAP)1 and c-IAP2. DHEA also caused a significant decrease of transforming growth factor-β1-induced collagen production and fibroblast to myofibroblast differentiation, and inhibited platelet-derived growth factor-induced fibroblast migration.
These findings demonstrate a disproportionate decrease of DHEA/DHEA-S in IPF patients and indicate that this molecule has multiple antifibrotic properties.
Abstract
DHEA/DHEA-S is abnormally decreased in IPF patients and this adrenal steroid has strong antifibrotic effects in vitro http://ow.ly/nD9sz
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic lung disease of unknown aetiology that causes progressive and irreversible destruction of the lung architecture [1, 2]. The disease has a dismal prognosis, with a median survival of ∼2–3 years after diagnosis [3]. Patients with IPF are usually aged 50–70 years and disease frequency increases markedly with age [4]. Some studies indicate that, at least in part, the ageing association can be related to an accelerated shortening of telomeres [5]. However, the mechanisms linking ageing with IPF are largely unknown [1, 6].
Dehydroepiandrosterone (DHEA) and its sulfate ester (DHEA-S) are the most abundant adrenal steroids in humans [7]. Blood levels of DHEA/DHEA-S reach a peak between the ages of 25 and 30 years and thereafter gradually decline, so that, by the age of 60, the concentrations are only 10–20% of corresponding values in young adults [7].
Abnormally decreased DHEA has been linked with immunosenescence, a decline of the immune system that occurs with ageing [8], as well as with several chronic diseases also associated with the elderly, e.g. insulin resistance, atherosclerosis, systemic arterial hypertension and Sjögren syndrome [9–11]. Accordingly, DHEA has been proposed as an ageing-protective molecule.
In this study, we analysed the plasma and bronchoalveolar lavage (BAL) levels of DHEA and DHEA-S in patients with IPF and age-matched controls. We further examined whether DHEA could affect the proliferation, migration and apoptosis of human lung fibroblasts, as well as the differentiation of fibroblasts to myofibroblasts and collagen synthesis. Our results show that DHEA/DHEA-S is abnormally decreased in IPF patients and that this adrenal steroid has strong antifibrotic effects in vitro.
Materials and methods
Study population
Plasma samples were obtained from 137 consecutive IPF patients, 87 males and 50 females (aged 64.1±9.9 years) diagnosed at the National Institute of Respiratory Diseases (INER), Mexico City, Mexico, and from 58 age- and sex-matched controls. The healthy donors, 30 males and 28 females (aged 56.5±4.6 years) were enrolled from the personnel of the INER and from our smoking cessation programme, according to their willingness to participate in the study. The Bioethics Committee of the INER approved the protocol, and signed consent was obtained from patients and controls.
BAL was carried out as previously described [12] in three control donors (aged 58±7.7 years) and 12 IPF patients (aged 62.8±4.6 years), all males.
Control subjects underwent a brief clinical history to exclude the presence of other disorders, such as diabetes, cardiovascular disease, endocrine disorders or cancer. Diagnosis of IPF was confirmed according to American Thoracic Society/European Respiratory Society guidelines [2].
DHEA/DHEA-S analysis
Plasma and BAL levels of DHEA were measured by using a solid-phase 125I radioimmunoassay (DHEA RIA DSL 9000; Siemens Medical Solutions Diagnostics, Los Angeles, CA, USA) and DHEA-S by chemiluminescent assay (DHEA SC chemiluminescent LKDS-1; Siemens Medical Solutions Diagnostics).
Cell culture
Human normal lung fibroblasts, CCD25-Lu, were acquired from the American Type Culture Collection. Fibroblast primary cell lines from normal and IPF lungs were obtained as described [13]. CCD25-Lu were cultured in DMEM while primary cell lines were cultured in Ham's F-12 medium all of them supplemented with 10% fetal bovine serum, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. Cell cultures were maintained at 37°C in a humidified atmosphere of 5% carbon dioxide. Cell viability was assessed by trypan blue exclusion.
Growth rate assay
Cell number was assessed by tetrazolium colorimetric WST1 assay (Roche, Mannheim, Germany), as described [13]. Fibroblasts were plated into 96-well microplates (10 000 cells·cm−2) and cultured with or without DHEA at concentrations of 50, 100 and 200 μM for 24, 48 and 72 h. At the end of the experiment, the medium was replaced by fresh medium containing WST1 solution and absorbance was analysed on an ELISA plate reader (Sinergy HT, BioTek, Winooski, VT, USA). The effect of DHEA on growth rate was expressed as a percentage of the control. These DHEA concentrations have been used previously [14–16].
Bromodeoxyuridine incorporation
Cells were seeded at a density of 10 000 cells·cm−2 in 96-well microplates, allowed to attach overnight and treated with or without DHEA (50, 100 and 200 μM) for 24 and 48 h. Fibroblast proliferation was evaluated by analysing 5′-bromo-2′-deoxyuridine (BrdU) incorporation into newly synthesised DNA using a cell proliferation ELISA (Roche) [17]. Optical density was measured using an ELISA plate reader (Sinergy HT, BioTek) at 450/595 nm. Proliferation was expressed as a percentage of control.
Apoptosis assay
Apoptosis was examined by flow cytometry using dual staining with fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodine (Annexin staining kit; BD Biosciences, San Jose, CA, USA) according to the manufacturer's instructions, as previously described [18]. Briefly, 1×106 cells in 100 μL of annexin buffer were stained with propidium iodide staining solution and annexin V-FITC staining solution. Cells were incubated at room temperature in the dark for 15 min and data acquired using a FACSAria flow cytometer (Becton-Dickinson, San Jose, CA, USA). Data were analysed using FlowJo 8.7 software (Tree Star, Inc., Ashland, OR, USA).
Caspase activity
Fibroblasts were lysed in caspase buffer (10 mM Tris-HCl, 10 mM NaH2PO4, 130 mM NaCl, 1% (v/v) Triton X-100). Protein concentration was assessed by the Bradford method, and caspase activity was assayed using a fluorometric method as previously reported [19], in a luminescence spectrometer (Sinergy HT, BioTek) at 380/430–460 nm. Acetyl tetrapeptides coupled to 4-methycoumaryl-7-amide were used as substrates: Ac-DEVD-AMC (caspase-3/7/8) and Ac-LEHD-AMC(caspase-9) (PeptaNova GmdH, Sandhausen, Germany). Results are expressed as the change in unit of fluorescence (UF)·min−1·mg−1 protein produced after substrate cleavage.
Human apoptosis antibody array
The expression profile of 35 apoptosis-related proteins was measured using a human apoptosis array kit (Proteome Profiler Array ARY009; R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions as described [20]. Mean spot pixels were quantified by densitometry. Data were expressed as fold-change and arbitrarily considered as biologically important at a change >30%.
Migration assay
Fibroblast migration was assayed using 24-well collagen-coated Boyden chambers (Chemicon, Millipore, Bedford, MA, USA) with an 8-μm pore size as described [21]. Cells (2×105) were seeded in the upper chamber and DHEA (100 μM), platelet-derived growth factor (PDGF) 25 ng·mL−1 (R&D Systems) or both were added in the lower chamber containing 0.5 mL of medium with 5% bovine serum albumin (BSA). After incubation for 8 h the migrating cells were quantified according to the manufacturer’s instructions.
Western blotting
Fibroblasts were lysed with RIPA buffer (Sigma Aldrich, St Louis, MO, USA) containing multiple protease inhibitors (Calbiochem, Merck KGaA, Darmstadt, Germany). Sample proteins (40 μg) were separated by 10% SDS-PAGE electrophoresis, transferred onto nitrocellulose membranes (Bio Rad, Hercules, CA, USA) and blocked following standard protocols. Membranes were incubated overnight at 4°C with primary antibodies: α-smooth muscle actin (SMA) (1:500; Sigma Aldrich), caspase-9 (1:500; Abcam, Cambridge, MA, USA) and β-tubulin (1:500; Santa Cruz Biotechnology Inc., Dallas, TX, USA). Incubation with secondary antibodies coupled to horseradish peroxidase (anti-mouse and anti-rabbit IgG; Invitrogen Life Technologies, Grand Island, NY, USA) was performed for 1 h at room temperature and the signal was visualised with the Super Signal West Pico Enhanced Chemiluminescence detection system (Thermo Scientific, Rockford, IL, USA). Signals were normalised against β-tubulin.
Immunofluorescence
Fibroblasts were plated on coverslips and incubated for 48 h with serum-free medium alone or containing 100 μM DHEA, 5 ng·mL−1 transforming growth factor (TGF)-β1 or both. Cells were fixed with acetone/methanol at 4°C, permeabilised with 0.5% Triton X-100 and incubated with human monoclonal α-SMA antibody (Sigma Aldrich) at 4°C overnight in a humidified chamber. Cells were then treated with an affinity-purified goat anti-mouse IgG DyLight 549 (Biocare Medical, Concord, CA, USA) for 1 h in a humidified chamber at room temperature. Slides were counterstained with 4,6-diamidino-2-phenylindole (DAPI) mounting medium (Santa Cruz Biotechnology Inc.).
Collagen assay
Fibroblasts were seeded into 25-cm2 culture flasks and cultured until 70% confluence and stimulated in serum-free medium with DHEA (100 μM), TGF-β1 (5 ng·mL−1) or both for 48 h. Supernatants were dialysed, lyophilised and resuspended to a volume of 100 μL. Collagen concentration was determined by colorimetric Sircol soluble collagen assay (Biocolor Ltd., Carrickfergus, UK) according to the manufacturer's instructions [22]. Optical density was measured at 555 nm using an ELISA plate reader (Sinergy HT, BioTek). Collagen concentration was expressed as relative to control.
Statistical analysis
Plasma level results were not normally distributed and were logarithmically transformed for statistical analysis. Since the study groups did not follow the normal distribution, we used a nonparametric approach for the descriptive statistics (median and range) and the statistical analysis (Mann–Whitney U-test). In vitro data are given as mean±sd. Differences were assessed by one-way ANOVA or Wilcoxon test. p<0.05 was considered significant. Data were analysed using the statistical program SPSS for Windows, release 14.0 (SPSS Inc., Chicago, IL, USA).
Results
DHEA and DHEA-S are decreased in patients with IPF
Plasma levels of DHEA and DHEA-S were measured by radioimmunoassay and chemiluminescent assays, respectively, in 137 IPF patients and 58 controls. As illustrated in figure 1, both molecules were significantly decreased in male IPF patients (median (range) DHEA: 4.4 (0.2–29.2) versus 6.7 (2.1–15.2) ng·mL−1, p<0.01; DHEA-S: 47 (15.0–211) versus 85.2 (37.6–247.0) μg·dL−1, p<0.001), while in females only DHEA-S was significantly decreased (32.6 (15.0–303.0) versus 68.3 (16.4–171) μg·dL−1, p<0.001). DHEA in females showed a nonsignificant tendency to decrease (4.3 (0.9–28.9) versus 5.7 (0.98–18.3) ng·mL−1, p=0.2).

Sulfated dehydroepiandrosterone (DHEA-S) is decreased in patients with idiopathic pulmonary fibrosis (IPF). Plasma levels of a) dehydroepiandrosterone (DHEA) and b) DHEA-S were examined by radioimmunoassay and chemiluminescent assay in male and female healthy donors and IPF patients. The line in each box represents the median and boxes the 25th and 75th percentiles. The whiskers represent 5th and 95th percentiles. The open triangles and circles represent outliers. **: p<0.01; ***: p<0.001.
DHEA-S concentration was also significantly decreased in BAL samples from IPF patients (2.31±0.61 versus 1.37±0.14 μg·dL−1; p<0.01). No differences were observed in DHEA levels.
No correlations were found between clinical and functional parameters (e.g. forced vital capacity) and the DHEA or DHEA-S plasma concentrations (data not shown).
DHEA decreases lung fibroblast proliferation
Cell growth was quantified by the WST1 proliferation assay in the CCD25-Lu normal cell line as well as in three primary cell lines, one obtained from normal lungs and two from IPF lungs. No differences in growth rate were observed under basal conditions between IPF and control fibroblasts (152±10% versus 157±7% at 48 h and 214.0±2.99% versus 200.4±10.1% at 72 h). As shown in figure 2, DHEA treatment caused a dose- and time-dependent decrease of cell growth. In general, this effect was significant with DHEA concentrations ≥50 μM and was stronger after 48 h. The inhibitory effect of DHEA on human lung fibroblast proliferation was corroborated by measuring BrdU incorporation into DNA (fig. 2e and f). In normal and IPF fibroblasts, DHEA caused a significant decrease in cell proliferation at concentrations from 50 μM.
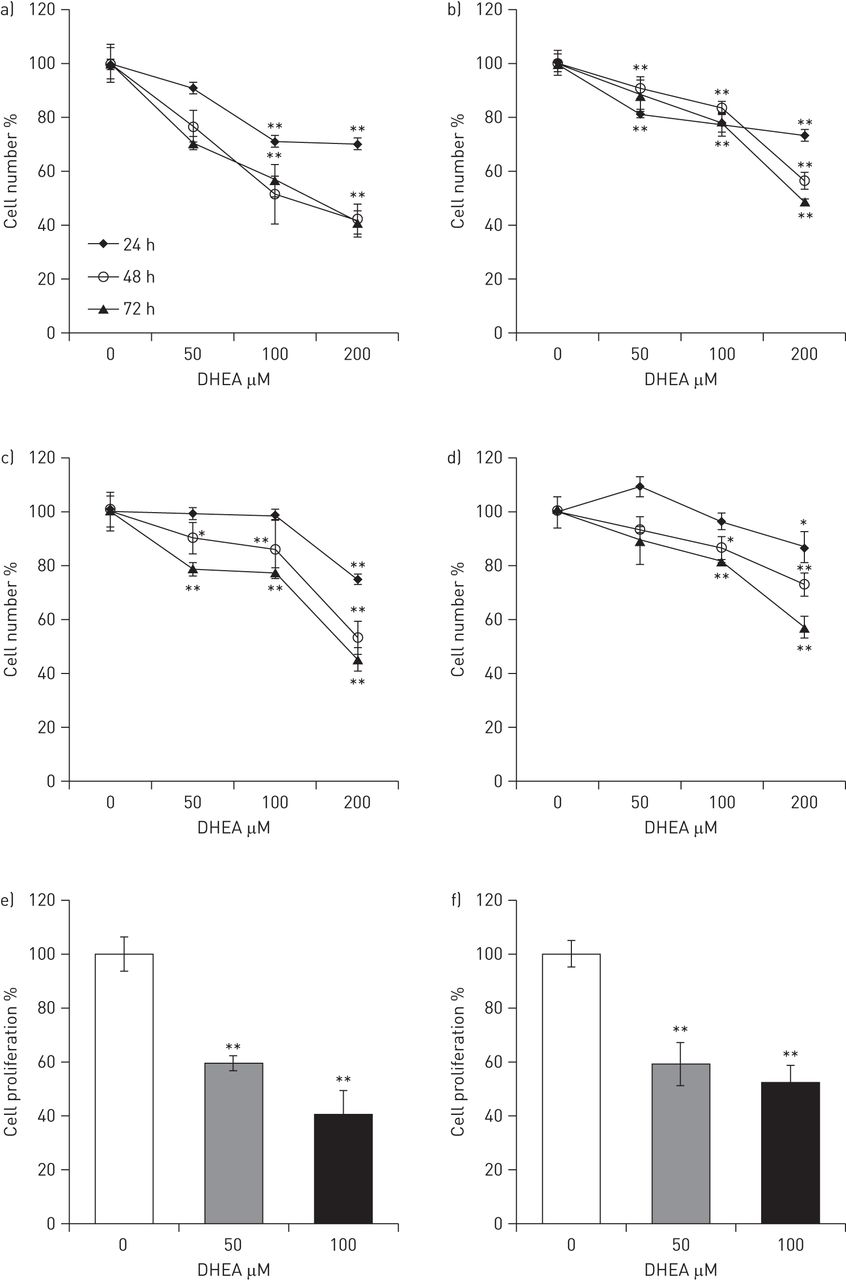
Dehydroepiandrosterone (DHEA) decreases growth rate and proliferation in normal and idiopathic pulmonary fibrosis (IPF) human lung fibroblasts. Growth rate was measured by WST1 assay in two normal (a and b) and two IPF (c and d) human lung fibroblast cell lines at 24, 48 and 72 h in the absence or presence of increasing concentrations of DHEA (50, 100 and 200 μM). Cell proliferation was measured by 5′-bromo-2′-deoxyuridine (BrdU) incorporation into DNA at 24 h in e) a normal and f) an IPF fibroblast cell line in the absence or presence of increasing concentrations of DHEA (50 and 100 μM). Data are expressed as mean±sd from three independent experiments. *: p<0.05; **: p<0.01.
DHEA induces apoptosis in lung fibroblasts
The effect of DHEA on cell death was examined using concentrations of 100 and 200 μM. To detect early apoptosis, DHEA-treated and control fibroblasts were assayed for the translocation of phosphatidylserine from the inner to the outer leaflet of the plasma membrane using the annexin V binding assay. As shown in figure 3a, at 24 and 48 h of culture both DHEA concentrations provoked a significant increase in fibroblast apoptosis. To examine whether the apoptosis induced by DHEA was mediated by the extrinsic or intrinsic pathway we evaluated caspase-8 and caspase-9 by immunoblotting. Stimulation with 100 μM DHEA induced an increase of active caspase-9 in lung fibroblasts at 24 h (fig. 3b). Activation of pro-caspase-9 was corroborated by fluorometry (fig. 3c). No changes were observed with pro-caspase-8 (data not shown). We examined whether DHEA induced apoptosis in IPF fibroblasts and if it was influenced by the inhibition of caspase-9. We found that basal apoptosis (2.8±1.3%) increased to 6.1±0.6% when cells were exposed to 100 μM DHEA (p<0.05). Caspase-9 inhibitor attenuated DHEA-induced fibroblast apoptosis (3.6±0.3%; p<0.05).

Dehydroepiandrosterone (DHEA) induces apoptosis of human lung fibroblasts. a) Normal fibroblast cell line CCD25-Lu was incubated for 12, 24 and 48 h in the absence or presence of increasing concentrations of DHEA (100 and 200 μM) in fetal bovine serum supplemented medium. Apoptosis and necrosis were quantified by fluorescence-activated cell sorter using fluorescein isothiocyanate-conjugated annexin V and propidium iodide, respectively. Levels of apoptosis are expressed as percentage of the total number of acquired events in at least three independent experiments. Data are expressed as mean±sd. **: p<0.01. b) Representative western blot analysis of pro-caspase-9 and active caspase-9 expression in total lysates obtained from the normal human lung fibroblast cell line CCD25-Lu. Fibroblasts were exposed to 100 μM DHEA for 0, 4, 12 and 24 h, and β-tubulin was used as loading control to normalise by densitometric analysis. c) Caspase-9 activity was also assayed by a fluorometric method using Ac-LEHD-AMC as a substrate in lysates of normal cells exposed for 12 and 24 h to medium with or without 100 μM DHEA. Data are expressed as mean±sd from three independent experiments. UF: units of fluorescence.
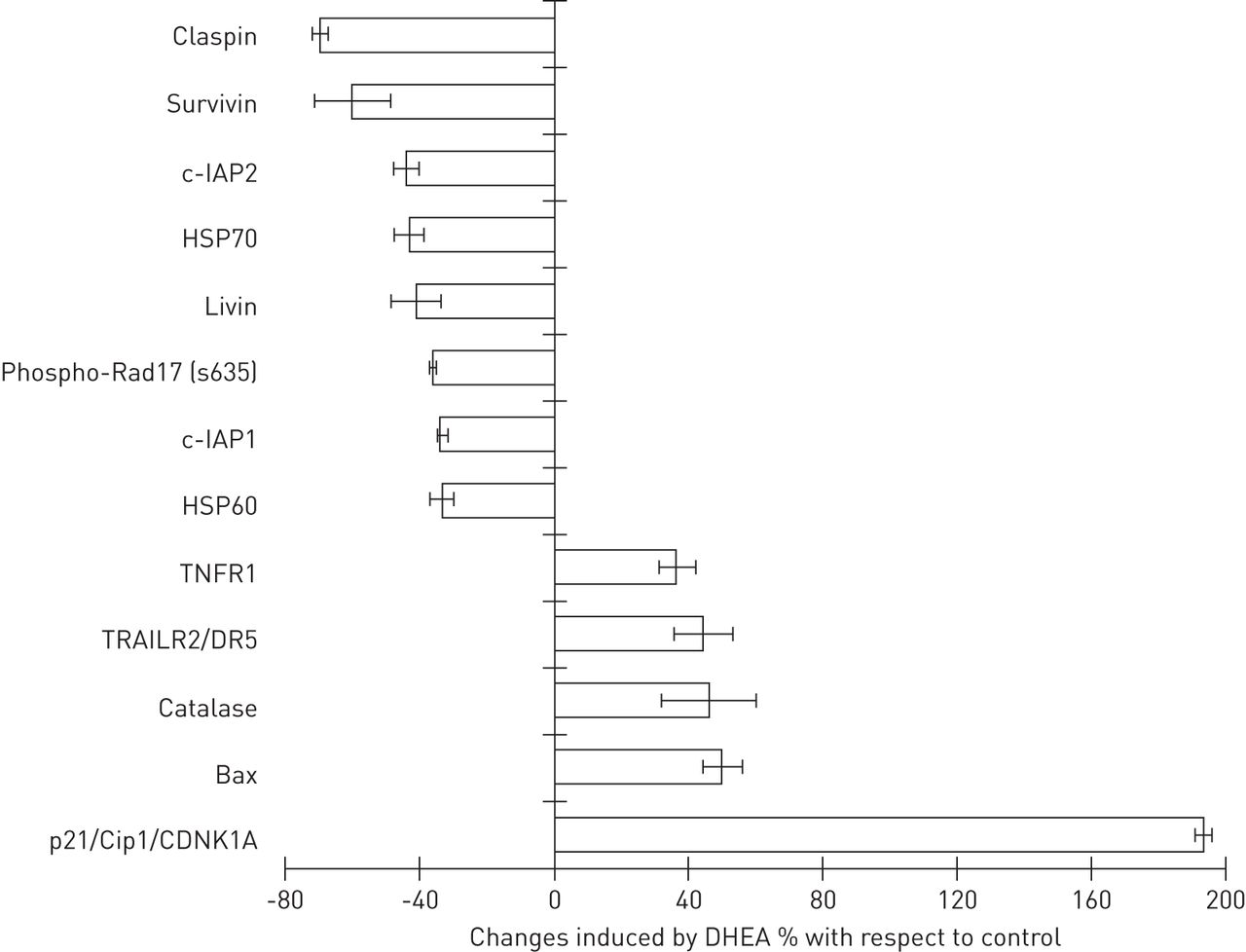
To further explore the apoptotic/anti-apototic proteins likely to be involved in DHEA-induced cell death, we used a human apoptosis antibody array (fig. 4). Our results showed that DHEA increased some apoptosis receptors, such as tumour necrosis factor receptor 1 (TNFR1) (36.5±5.2%) and TRAILR2 (43.9±8.7%). An increase in levels of the pro-apoptotic marker Bax (49.7±6%) and of the cyclin-dependent kinase inhibitor CDNK1A (p21/Cip1/CDNK1A), which inhibits cell cycle progression [23], was also detected. In addition, DHEA induced a decrease in the levels of several anti-apoptotic proteins, such as the cellular inhibitor of apoptosis c-IAP1 (34±1.5%), c-IAP2 (44.4±3.7%), survivin (60.5±11%), claspin (70±2.2%) and livin (41.6±7%).

Effect of dehydroepiandrosterone (DHEA) on proteins involved in apoptosis. The expression profile of 35 apoptosis-related proteins was measured using a human apoptosis array kit as described in the Methods. Normal fibroblasts CCD25-Lu were treated with or without 100 μM DHEA for 24 h and cell lysates were used for antibody array. Data are expressed as per cent change±sd with respect to the untreated control. Data are shown for those proteins where changes >30% were observed. c-IAP: cellular inhibitor of apoptosis; HSP: heat shock protein; TNFR1: tumour necrosis factor (TNF) receptor 1; TRAILR2: TNF-related apoptosis-inducing ligand receptor 2; CDNK: cyclin-dependent kinase inhibitor.
DHEA inhibits PDGF-induced migration of lung fibroblasts
The effect of DHEA on cell migration was examined using collagen-coated Boyden chambers in two normal (fig. 5a and b) and two IPF (fig. 5c and d) fibroblast cell lines. The number of cells that migrated in the presence of medium plus 5% BSA alone was used as control, while PDGF, a potent fibroblast chemoattractant, was used as a positive control. As shown in figure 5, PDGF-induced fibroblast migration was significantly reduced by DHEA in both normal human lung and IPF fibroblasts. Around 30–70% of PDGF-BB-induced fibroblast migration was inhibited by 100 μM DHEA.

Dehydroepiandrosterone (DHEA) inhibits platelet-derived growth factor (PDGF)-induced migration of lung fibroblasts. Cell migration assay was performed in normal (a and b) and idiopathic pulmonary fibrosis (c and d) lung fibroblasts with incubation for 8 h in the absence (control) or presence of 100 μM DHEA and/or 25 ng·mL−1 PDGF-BB in collagen-covered Boyden chambers as described in the Methods. Data are expressed as mean±sd from three independent experiments. *: p<0.05; **: p<0.01.
DHEA inhibits TGF-β1-induced fibroblast to myofibroblast differentiation
The effect of DHEA on the expression of α-SMA, the hallmark of myofibroblasts, was examined by Western blot using two different approaches. In the first approach, fibroblasts were cultured simultaneously over 48 h with TGF-β1 and DHEA. As exemplified in figure 6, 100 μM DHEA concentrations induced a strong reduction in TGF-β1-induced α-SMA in both normal (fig. 6a and b) and IPF (fig. 6c and d) fibroblasts. In the second approach, fibroblasts were first stimulated with TGF-β1 for 48 h, and then treated with DHEA for an additional 48 h, without removing the growth factor. As shown in figure 6e, an inhibitory effect was observed with 100 μM DHEA in normal human lung fibroblasts. The attenuation of fibroblast to myofibroblast differentiation by DHEA was corroborated by immunofluorescence, both in normal lung fibroblasts (figure 7a–h) and IPF fibroblasts (figure 7i–p).

Dehydroepiandrosterone (DHEA) inhibits fibroblast to myofibroblast differentiation induced by transforming growth factor (TGF)-β1. Representative Western blots of α-smooth muscle actin (α-SMA) in total lysates of fibroblasts that were exposed to 5 ng·mL−1 of TGF-β1 and/or to DHEA in fetal bovine serum supplemented medium for 48 h. Normal (a and b) and idiopathic pulmonary fibrosis fibroblasts (c and d) were stimulated simultaneously with TGF-β1 and 100 μM DHEA. Densitometric analysis of each experiment is included in the right panel. DHEA caused a two- to four-fold decrease in TGF-β1 induced α-SMA expression (p<0.05). e) Normal fibroblasts were stimulated with TGF-β1 for 48 h and subsequently with increasing concentrations of DHEA (25, 50 and 100 μM) for an additional period of 48 h without removing TGF-β1. β-tubulin content was used as a loading control for normalisation.

Dehydroepiandrosterone (DHEA) inhibits α-smooth muscle actin (α-SMA) expression induced by transforming growth factor (TGF)-β1 in normal (a–h) and idiopathic pulmonary fibrosis (IPF) (i–p) lung fibroblasts. Representative immunofluorescence staining for α-SMA from human normal (a–d) and IPF (i–l) fibroblasts stimulated with or without 5 ng·mL−1 TGF-β1 and/or 100 μM DHEA for 48 h. Simultaneous staining of nuclei with 4,6-diamidino-2-phenylindole (DAPI) is also shown in blue (e–h, m–p). Scale bars=40 μm.
DHEA reduces TGF-β1-induced collagen production
To investigate whether DHEA has an effect on collagen synthesis, human lung fibroblasts were stimulated with TGF-β1 and collagen protein was measured in cell culture supernatants. As shown in figure 8, collagen production increased between two- and three-fold upon stimulation with TGF-β1 as assessed by the Sircol assay. This effect was significantly reduced by DHEA at 100 μM in both normal human lung fibroblasts (fig. 8a and b; p<0.01) and IPF-derived fibroblasts (fig. 8c and d; p≤ 0.05).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dehydroepiandrosterone (DHEA) reduces transforming growth factor (TGF)-β1-induced collagen production. Normal (a and b) and idiopathic pulmonary fibrosis (c and d) lung fibroblasts were treated with or without 5 ng·mL−1 TGF-β1 and/or 100 μM DHEA for 48 h. Collagen concentration in conditioned media was quantified by Sircol collagen soluble assay. Data are expressed as mean±sd from three independent experiments. *: p≤0.05; **: p<0.01.
Discussion
IPF is a progressive and usually lethal lung disease of unknown aetiology occurring primarily in elderly adults [1, 4, 6]. However, the mechanisms that link ageing with IPF remain elusive, although accelerated shortening of telomeres that may affect alveolar epithelial cell regeneration, and epigenetic processes that seem to compromise both epithelial cells and fibroblasts have been proposed [1, 5, 6, 24].
DHEA and DHEA-S are anabolic pro-hormones secreted by the human adrenal cortex. DHEA-S is the hydrophilic storage form that circulates in the blood while evidence indicates that lipophilic DHEA is the form that is converted intracellularly into androgens and oestrogens. Circulating concentrations of DHEA and DHEA-S decline progressively throughout adult life, so that after 60–70 years of age the levels are only 10–20% of those detected in young adults.
Lower circulating levels have been correlated with the incidence of inflammatory and malignant processes [25, 26]. Thus, blood DHEA-S level has been found independently and inversely associated with immunosenescence, autoimmune diseases, non-alcoholic steatohepatitis, insulin resistance, atherosclerosis and systemic arterial hypertension [8–11, 27]. Evidence indicates that low levels of DHEA-S predict death from all causes, cardiovascular disease and ischaemic heart disease in older males [28]. Moreover, it has recently been shown that long-term DHEA replacement may reduce insulin resistance and improve glucose tolerance in elderly, overweight or obese females and males with abnormal glucose tolerance [29].
In our study we found, for the first time to our knowledge, that IPF patients had a disproportionate decrease in the circulating levels of DHEA-S compared with age-matched controls. Studies in other fibrotic disorders are scarce, although more advanced non-alcoholic fatty liver disease, as indicated by the presence of severe fibrosis, is also strongly associated with low levels of circulating DHEA-S [26, 30]. However, the underlying mechanisms associated with the putative protective, antifibrotic role of DHEA are unknown.
In this context, it is important to emphasise that studies dealing with the mechanisms by which DHEA exerts its effects, primarily in fibroblasts, are scarce and have shown contradictory results. For example, regarding collagen synthesis, it has been reported that DHEA induced a modest increase in collagen gene expression in skin fibroblasts, but without any effect on collagen synthesis [31]. By contrast, and similar to our results, treatment with DHEA resulted in a significant decrease in procollagen type I mRNA and in procollagen type I protein synthesis in cardiac fibroblasts [32].
Regarding apoptosis, results in the literature are also contradictory. Thus, while DHEA was reported to have a protective role against apoptosis in immortalised human keratinocytes and neurons [33, 34], in other cell types, such as human hepatocellular carcinoma cells and pulmonary artery smooth muscle cells, DHEA induced apoptosis [15, 35]. Therefore, it is likely that the cellular response to DHEA is related to the cell type and/or the cell tissue origin.
In the present study, we evaluated the effects of DHEA on some important lung fibrogenic processes, including migration, proliferation, apoptosis and collagen synthesis by human lung fibroblasts, as well as on fibroblast to myofibroblast differentiation. DHEA was used because DHEA-S is the hydrophilic storage form that circulates in the blood, while DHEA is the functional form and the principal form used in steroid hormone synthesis. For this reason, despite the fact that DHEA-S is the form usually found to have decreased in blood studies in different chronic diseases associated to ageing, it is DHEA which is used for in vitro or in vivo experiments [36].
Fibroblast migration to the injured areas in response to chemoattractant signalling molecules, and their subsequent expansion in the lesion, are two important profibrotic mechanisms. In this context, our results showed that DHEA is a potent inhibitor of PDGF-induced migration of fibroblasts through a type 1 collagen matrix as well as on fibroblast proliferation, although the specific pathways that promote these activities remain to be determined.
In the fibroblastic foci of IPF lungs, fibroblasts undergo a phenotypic change to myofibroblasts with the subsequent upregulation of extracellular matrix macromolecules [1]. This process plays a key fibrogenic role in this disease where myofibroblasts persist and contribute to pathological scar formation. In this regard, we found that TGF-β1-induced fibroblast to myofibroblast differentiation, evaluated through the expression of α-SMA, was significantly reduced in a dose-dependent manner by DHEA. Attenuation of myofibroblast differentiation was also observed by immunofluorescence in IPF-derived fibroblasts.
Fibrotic disorders such IPF are characterised by increased deposition of fibrillar collagens, and we assumed that DHEA would suppress collagen synthesis by lung fibroblasts. Paralleling the inhibitory effect on myofibroblast differentiation, we observed that DHEA repressed TGF-β1-induced collagen synthesis.
The reasons why fibroblasts and myofibroblasts persist in the IPF fibroblastic foci and appear to be resistant to apoptosis are unclear. TGF-β1 plays an important role, promoting two myofibroblast prosurvival signalling pathways, focal adhesion kinase and protein kinase B [37, 38]. Therefore, we next examined the effect of DHEA on fibroblast cell death. Using fluorescence-activated cell sorter analysis to quantify the level of apoptosis, we observed that DHEA promotes fibroblast cell death, in a time-dependent manner. Importantly, our findings also showed that treatment with DHEA resulted in activation of caspase-9, indicating that DHEA activates the intrinsic pathway of apoptosis. Progression through the intrinsic pathway leads to mitochondrial release of cytochrome C into the cytosol, inducing apoptosome formation with the activation of procaspase-9, which in turn cleaves, and thereby activates, downstream effector caspases [39–41]. To identify other molecules likely to be involved in the DHEA-induced changes to the fine balance of pro-apoptotic and anti-apoptotic factors, we employed a human apoptosis antibody array. This array allowed semi-quantitative analysis of the changes occurring in critical apoptosis and checkpoint pathway proteins. Important anti-apoptotic proteins (c-IAP1, c-IAP2, livin, survivin, claspin) were downregulated, further supporting the evidence that the intrinsic apoptotic pathway is activated, while the increase in death receptors (TNFR1 and TRAILR2) suggests crosstalk between intrinsic and extrinsic apoptotic pathways.
All these findings on normal and IPF fibroblasts provide strong evidence that DHEA could act as an antifibrotic compound against fibroproliferative diseases, including, but not limited to, pulmonary fibrosis, although the intracellular mechanisms involved in these different biological effects need to be explored.
In this context, it is important to emphasise that the cell receptors and intracellular signalling pathways by which DHEA exerts its effects are presently unclear. Moreover, since DHEA is a multifunctional steroid with a broad range of biological effects, molecular mechanisms may be varied. Although DHEA binding to membrane-associated proteins has been demonstrated, definitive evidence for a DHEA-specific membrane receptor including isolation and sequencing of the gene for the purported receptor is lacking [42]. It has been hypothesised that DHEA may exert its effects through the oestrogen/androgen receptors. However, other studies have shown that the effects of DHEA on proliferation and apoptosis are independent of both receptors [43, 44], whereas some studies have implicated one receptor potentially coupled to a G-protein [42, 43]. In the past 10 years, a series of orphan receptors, namely the peroxisome proliferators activated receptor, pregnane X receptor and constitutive androstanol receptor, have been reported and there is evidence that DHEA (or some of its metabolites) either bind to or activate these newly characterised receptors [36].
Regarding signalling pathways, it has been demonstrated that DHEA affects the cell cycle by blocking the phosphatidylinositol 3-kinase pathway [15]. There is also evidence that DHEA negatively regulates the mitogen-activated protein kinase (MAPK) pathway in humans, via a novel MAPK phosphatase, suppressing the p38-MAPK cascade [45]. Additionally, in an experimental model of pulmonary hypertension, DHEA prevented the activation of the RhoA–ROCK (Rho kinase) signalling pathway, although the cellular target was not identified [46].
In summary, our findings demonstrate that the circulating level of DHEA/DHEA-S is decreased in IPF and indicate that DHEA displays a strong antifibrotic effect on fibroblasts, affecting migration, proliferation, differentiation to myofibroblasts, collagen synthesis and survival. These findings may open new therapeutic options for this destructive lung disease.
Footnotes
Support statement: This work was partially supported by Conacyt grant Salud-2009-01-113590.
Conflict of interest: None declared.
- Received February 14, 2012.
- Accepted October 11, 2012.
- ©ERS 2013
References










