





Abstract
The aim of this study was to characterise lung function and bronchoalveolar lavage sphingolipid profile in newborn mice during hyperoxia exposure and recovery in room air, and to examine the effect of d-sphingosine supplementation during recovery.
Newborn mice were exposed to 80% oxygen for 4 weeks and allowed to recover in room air for another 4 weeks. Lung function measurements and morphometrical analysis of lung tissue were performed and bronchoalveolar lavage fluid was collected during hyperoxia and recovery with and without d-sphingosine supplementation.
Hyperoxia exposure altered lung function, which partially recovered in room air. Lungs had fewer and enlarged alveoli which persisted during recovery. Multiple sphingolipids were significantly increased after hyperoxia. Ceramides were increased after 2 weeks of recovery, but normalised to control values after 4 weeks. The addition of d-sphingosine during the first 5 days of recovery accelerated the normalisation of ceramide levels at 2 weeks and partially reversed the hyperoxia-induced increase in alveolar size and arrest in alveolarisation at 4 weeks.
Exposure of newborn mice to hyperoxia caused restrictive and obstructive lung function changes that partially recovered in room air, while alveolar morphology remained abnormal. Hyperoxia increased ceramide levels, with normalisation after recovery. d-sphingosine addition during recovery reduced ceramide levels and ameliorated hyperoxia-induced alveolar arrest.
Abstract
d-sphingosine during recovery reduced ceramide levels and ameliorated hyperoxia-induced alveolar arrest in mice http://ow.ly/l3r8P
Introduction
Respiratory distress syndrome in preterm infants is often managed by mechanical ventilation and supplemental oxygen. This occurs in the setting of an immature, surfactant-deficient lung, devoid of antioxidant defenses. A subset of survivors develop bronchopulmonary dysplasia (BPD) [1], originally characterised by parenchymal fibrosis, oedema, vascular changes and persistent inflammation. Advances in neonatal care, including exogenous surfactant, prenatal steroids, better nutritional management and new modes of ventilation have improved the survival of infants with very low birthweight (VLBW). However, these VLBW infants are prone to develop “new BPD” with fewer and larger alveoli as a result of interrupted septation and abnormal vascular organisation [2]. Even though progress has been made in the management of infants with BPD, current treatment remains symptomatic.
Studies looking at the hyperoxia-induced model of BPD are numerous, but to this date no studies have looked at the influence of sphingolipids in this model. Sphingolipids are important structure-bearing constituents of the cell membrane which also function as regulatory molecules of cell proliferation and cell death, endothelial barrier function, angiogenesis and immune response [3]. Altered sphingolipid levels have been found in a variety of diseases, such as atherosclerosis, chronic heart failure [4], asthma [5], diabetes, sepsis, cystic fibrosis and chronic obstructive pulmonary disease [6]. Two sphingolipids, ceramide and sphingosine-1-phosphate, determine the pro- and anti-apoptotic balance [7]. In this rheostat, ceramide stimulates apoptosis and cell cycle arrest, and sphingosine-1-phosphate stimulates cell survival and proliferation. Increased apoptosis has been found in epithelial cells of BPD patients [8] and in animal models of BPD [9], and total lung ceramide levels are upregulated in hyperoxia-exposed neonatal rats [10]. To date, no detailed analysis of the sphingolipid metabolome in the hyperoxia-induced BPD model [11] has been reported. Furthermore, little is known about the evolution of lung injury in murine models of BPD. Mice exposed to >60% oxygen during the first days of life showed lung function abnormalities that persisted until 67 weeks of age [12], but the precise evolution of lung function during the acute phase of hyperoxic injury, followed by recovery in room air, has not been examined.
The objective of our study was to determine the evolution of lung function and sphingolipids in hyperoxia-exposed newborn mice during both the acute hyperoxic phase and recovery in room air. In addition, we studied the effect of pharmacological intervention on hyperoxia-induced lung damage by giving d-sphingosine supplementation during the recovery phase.
Materials and methods
Animals
C57/BL6 mice were obtained from Charles River (St Constant, Quebec, Canada) and animal studies were conducted according to criteria established by the Canadian Council on Animal Care and approved by the Animal Care and Use Committee of the Hospital for Sick Children (Toronto, Canada).
Hyperoxia-induced BPD
We used a modified hyperoxia model as described by Warner et al. [13]. Two pathogen-free timed pregnant C57/BL6 mice gave birth in room air. At post-natal day 1, the mothers and their pups were placed in paired Oxycycler exposure chambers (Biospherix Ltd, Lacona, NY, USA). Litters were exposed to hyperoxia (80% O2) or room air for 28 days. Litter sizes were kept at six pups in both the hyperoxia and room air groups. Dams were switched daily. Oxygen exposure was maintained for 28 days, after which the mice had gained enough weight to perform lung function measurements. Hyperoxia-treated litters were then exposed to room air for 7, 14 or 28 days. Room air control litters were treated in the same manner. d-sphingosine (Sigma-Aldrich, St Louis, MO, USA) was administered (1.25 μg·g−1 bodyweight, i.p.) daily to a subset of hyperoxia-treated litters during the first 5 days of recovery in room air. Control animals were treated with d-sphingosine in the same manner. At various times during these conditions, lung function was assessed, bronchoalveolar lavage (BAL) was performed, and tissues processed for histology (online supplementary fig. E1).
Lung function measurements
28 days after the start of hyperoxia, the flexiVent rodent ventilator (Scireq, Montreal, Canada) was used to assess lung function as described in detail in the online supplementary material. For the long-term follow-up experiments of hyperoxic mice in room air, mice were subjected to lung function measurements at postnatal day 35, 42 and 56.
BAL
Lungs were infused through the endotracheal tube with 600 μL sterile saline, followed by withdrawal and re-infusion two more times [14, 15]. The collected fluid was centrifuged at 1400×g for 8 min. The supernatant was collected in a siliconised Eppendorf tube and stored at -80°C for mass spectrometry analysis.
Measurement of sphingolipids
Sphingolipid levels in BAL fluid (BALF) were measured as described in the online supplementary material.
Histology and morphometry
Following lung function measurements, the lungs were pressure-inflated and processed for histology and morphometry, as described in the online supplementary material.
Statistics
All values are presented as mean±sem, assuming a normal distribution (SigmaPlot version 11 for Windows; Systat Software Inc., San Jose, CA, USA). Differences were assessed by t-test or, for comparison of three or more groups, by one-way ANOVA followed by Tukey’s honestly significant difference comparison. Significance was inferred as p<0.05.
Results
Lung function measurements
In newborn mice exposed to 28 days of hyperoxia, flexiVent measurements revealed a significant increase in resistance and reduced compliance, total lung capacity and forced vital capacity compared to controls (fig. 1a and table E1). Long-term follow-up of hyperoxia-exposed mice showed recovery of lung function parameters to control levels after 1 week of recovery in room air. Total lung capacity and static and dynamic compliance took 2 weeks to normalise (fig. 1b). However, forced expirations remained abnormal during the 4 weeks of recovery in room air. Flow–volume curves at 0 and 4 weeks of recovery showed persistent obstructive abnormalities with improvement of volume (fig. E2 and table E1). Addition of d-sphingosine during recovery in room air did not significantly improve lung function.

Lung function measurements a) after 4 weeks of hyperoxia exposure and b) during recovery in room air of hyperoxia-exposed mice and room-air controls. n = 12 per group for the 28-day time point, except for mean expiratory flow, where n = 8. n = 6 for the 35-, 42- and 56-day time points. Data are presented as mean±sem. *: p<0.05; ***: p<0.001.
Sphingolipid measurements
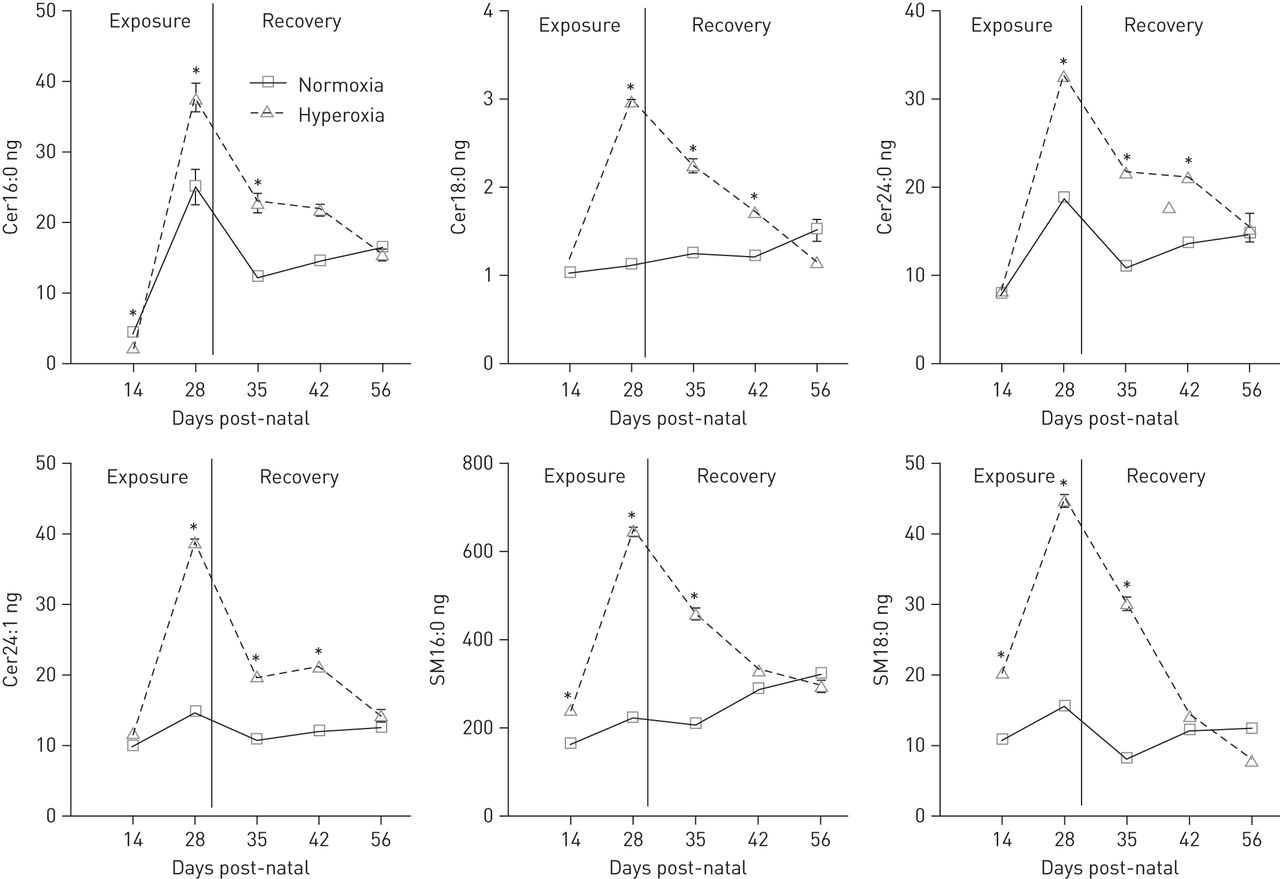
Sphingolipid levels in BALF measured by liquid chromatography with tandem mass spectrometry are shown in online supplementary tables E2–E4. A significant increase in multiple sphingomyelin and ceramide species was found after 2 and 4 weeks of hyperoxia exposure (fig. 2 and table E2). In particular, long chain ceramides (Cer16:0 and Cer18:0) and very long chain ceramides (Cer24:0 and Cer24:1) were greatly elevated (fig. 2 and table E2). Although small, significant increases were also found in sphinganine and a few dihydroceramides, both precursors of ceramides formed by the de novo pathway. All four major sphingomyelins (SM16:0, SM18:0, SM24:0 and SM24:1) showed two- to fourfold increases after hyperoxia exposure. Following 2 weeks of recovery in room air, all sphingomyelin species in the BALF of the hyperoxia-treated group returned to control levels, but many ceramide species were still significantly elevated compared with controls (fig. 2 and tables E3 and E4). Treatment with d-sphingosine accelerated the normalisation of ceramides in room air (fig. 3 and tables E3 and E4).
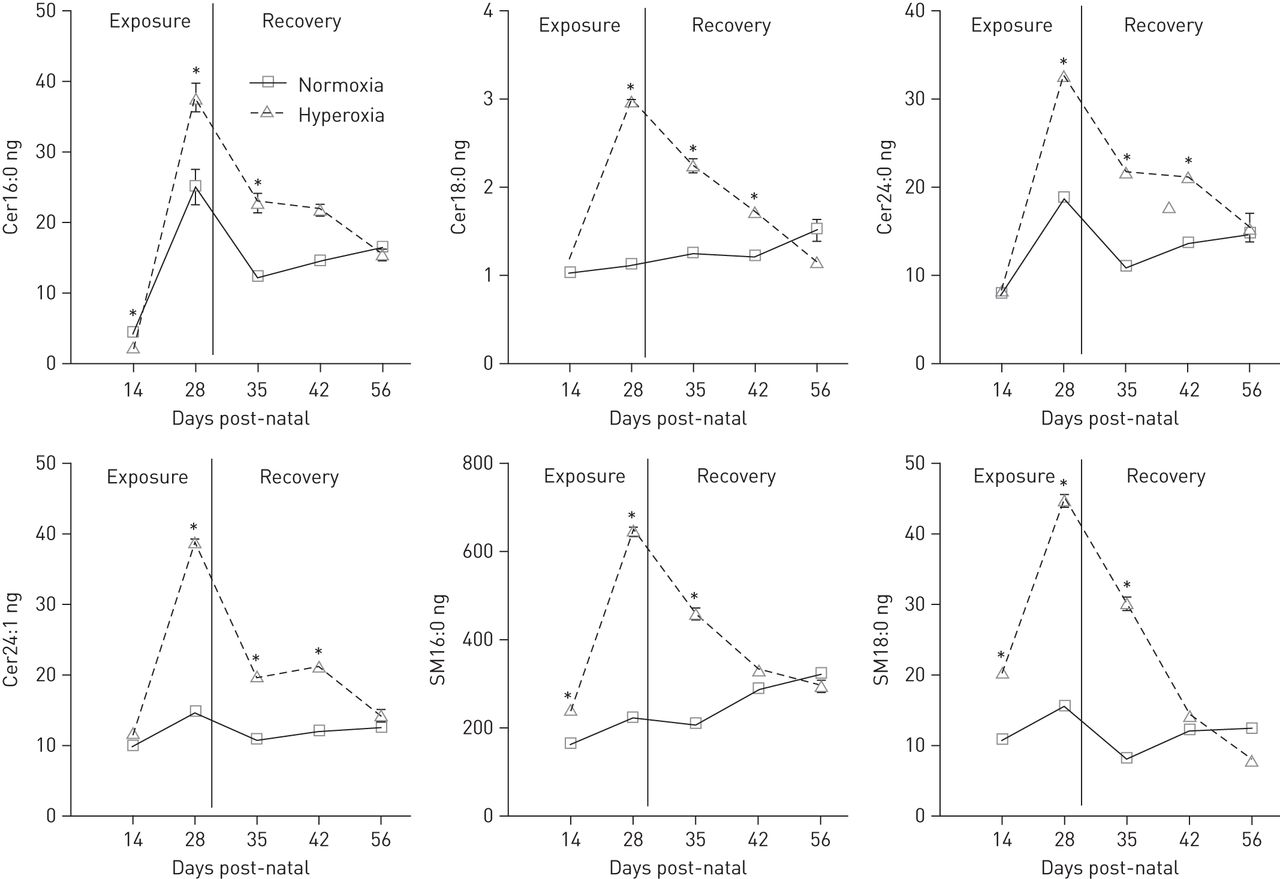
Representative sphingolipid measurements in total bronchoalveolar lavage fluid performed during hyperoxia treatment and recovery in room air. Results represent a total of 12 mice per group for the 14- and 28-day time points in hyperoxia and three mice per group for the 35-, 42- and 56-day time points of recovery in room air. Cer: ceramide: SM: sphingomyelin. *: p<0.05, when compared to room air-treated mice at the same time point.

Representative sphingolipid measurements in bronchoalveolar lavage fluid after 1 and 2 weeks’ recovery in room air with and without d-sphingosine (DS) treatment. Results represent a total of 12 mice per group for the 28-day time point in hyperoxia and three mice per group for the 35-, 42- and 56-day time points of recovery in room air and the DS supplementation experiments. Cer: ceramide; SM: sphingomyelin. *: p<0.05 in hyperoxia- and DS-treated mice when compared to normoxia- and DS-treated mice at the same time point; #: p<0.05 in hyperoxia-treated mice when compared to normoxia-treated mice at the same time point.
Histological analysis
Compared to room-air controls (fig. 4a), histological sections of hyperoxia-exposed lungs (fig. 4b) showed a homogeneous pattern of decreased alveolar septation, compared to room air-exposed newborn mice. Morphometry demonstrated a significant increase in mean linear intercept, a reduction in radial alveolar count and in the number of alveoli. Lung sections of mice exposed to 4 weeks of hyperoxia and allowed to recover in room air (fig. 4c) showed fewer and enlarged alveoli when compared to room-air controls. No significant difference in mean linear intercept and radial alveolar count was noted at 1, 2 and 4 weeks of recovery in room air (fig. 4e). Interestingly, when compared to hyperoxia-exposed nontreated mice (fig. 5a), mice treated with d-sphingosine for 5 days immediately after hyperoxia exposure (fig. 5b) had a significantly lower mean linear intercept and higher radial alveolar count (fig. 5c) at 2 weeks of recovery in room air. Mean linear intercept and radial alveolar counts of hyperoxia-exposed and d-sphingosine-treated mice did not return to control levels.

Effect of hyperoxia and subsequent room air recovery on lung histology. Representative histological sections of a) room air-exposed newborn mice; b) hyperoxia-treated newborn mice after 4 weeks of exposure; and c) hyperoxia-treated newborn mice that were allowed to recover for another 4 weeks in room air. Sections were stained with haematoxylin and eosin. Morphometry results are expressed as mean±sem for both room-air and hyperoxia groups for d) mean linear intercept (n = 8); e) radial alveolar counts (n = 8); and f) alveolar number (n = 4). Morphometry results for 4 weeks of recovery in room-air are expressed as mean±sem for g) mean linear intercept (n = 4) and h) radial alveolar count (n = 3). *: p<0.05; ***: p<0.001. Scale bars = 200 μm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of d-sphingosine supplementation on lung histology. Representative histological sections of lungs after 4 weeks of hyperoxia exposure followed by a) 2 weeks of recovery in room air and b) 2 weeks of recovery in room air with d-sphingosine (DS) supplementation. Sections were stained with haematoxylin and eosin. Scale bars = 200 μm (main) and 50 μm (inserts). c, d) Morphometry results represent a total of three mice per group and are expressed as mean±sem for all groups. *: p<0.05.
Discussion
In this study we examined BALF sphingolipid levels, lung function and histology in neonatal mice during hyperoxia exposure and after recovery in normoxia. Our findings showed that 4 weeks of exposure to hyperoxia causes alveolar damage and obstructive lung function abnormalities, together with increased sphingolipid levels, including ceramides. We demonstrated normalisation of ceramides and partial improvement of lung function, but found no improvement in histological abnormalities within 4 weeks of recovery in room air. d-sphingosine supplementation during normoxic recovery accelerated ceramide normalisation and improved hyperoxia-induced alveolar arrest, but did not affect lung function.
In our hyperoxia model, the increased airway resistance and decreased mean forced expiratory flow and forced expiratory volume could be caused by airway remodelling as a consequence of ongoing inflammation [13]. The observed changes in lung function are similar to those reported for newborn mice after 14 days of 85% oxygen exposure [16]. Another possible explanation for the lung function changes is a reduced number of alveolar attachments leading to decreased radial traction, which would result in airway narrowing and earlier airway collapse during expiration [17]. Prolonged exposure to hyperoxia in newborn mice caused an arrest in alveolarisation, thereby delaying the formation of secondary septa, reducing alveolar number and increasing alveolar wall thickness. In the present study, this decrease in alveolarisation was reflected in the radial alveolar counts.
At the end of 4 weeks of hyperoxia exposure, we observed a combined obstructive and restrictive pattern that shifted to an obstructive pattern at the end of 4 weeks of recovery in room air. The persistent abnormal forced expiration measurements, as well as the abnormal morphology, after 4 weeks in room air indicate incomplete recovery and a permanently altered lung structure. Velten et al. [16] studied newborn mice that were exposed to 85% oxygen for 14 days and recovered for 14 days in room air, and these newborns developed an increased resistance at 14 days, followed by an increased compliance of the lungs at 28 days. Yee et al. [12] noted a reduced compliance after 4 days of 100% oxygen exposure in newborn mice, and an increase in compliance at 67 weeks of age. They speculated that the transient decrease in compliance during the acute phase of hyperoxic injury was due to pulmonary oedema, impaired surfactant protein-C production and pro-inflammatory free oxygen radicals. Both studies seemed to indicate that the response of the newborn lung to hyperoxia can be divided into two stages: acute lung injury characterised by a less compliant lung, and chronic lung injury, characterised by a more compliant lung. Our measurements after 4 weeks of recovery could reflect an intermediary phase, between acute and long-term hyperoxia-induced lung injury, in which oedema and inflammation decreases and the lung becomes more compliant. We observed partial recovery of lung function, but no apparent recovery in morphology after 4 weeks of recovery in room air. This emphasises the importance of obtaining both structural and physiological data when tracking changes in disease state or monitoring the effects of an intervention. The persistence of morphological abnormalities in our mouse model is in line with the abnormalities seen in adult survivors of BPD [18, 19], and the persistent abnormal lung function parameters are consistent with results of lung function measurements performed in human survivors of BPD [20, 21], making this model a good choice for intervention studies aimed at repairing hyperoxia-induced lung damage.
Ceramides play an important role in apoptosis and lung inflammation [22, 23]. Kolliputti et al. [24] recently showed that ceramides regulate endothelial permeability and the inflammatory process caused by hyperoxia in adult mice. Ceramides have long been known to mediate acute lung injury by increasing alveolar permeability and pro-inflammatory cytokine production [25], but no studies have reported detailed ceramide profiles in neonatal hyperoxia-induced lung damage and during recovery in room air. Our study shows that multiple ceramide and sphingomyelin species are increased during exposure of neonatal pups to hyperoxia. Long chain ceramides (Cer16:0, Cer18:0 and Cer20:0) have antiproliferative and pro-apoptotic effects [26], whereas very long chain ceramides (Cer22:0, Cer24:0 and Cer24:1) promote cell proliferation [27]. We observed an eightfold increase in long-chain Cer16:0 during hyperoxia, suggesting a role for increased apoptosis via ceramide signalling in this hyperoxia model. Indeed, increased apoptosis has been observed in the lungs of mice and rats exposed to hyperoxia [28–30], similar to that noted in autopsy lungs of BPD patients [31]. Very long chain (Cer24:0 and Cer24:1) ceramides demonstrated a two- to threefold increase in our model, suggesting a role for proliferation, possibly of smooth muscle cells, which have been shown to proliferate in rat and mouse models of hyperoxia-induced BPD [12, 29, 32]. Both ceramides and sphingomyelin levels were increased, suggesting that the increase of ceramides is not due to breakdown of sphingomyelin via sphingomyelinase. The increased sphingosine levels (online supplementary table E2) may indicate increased ceramide formation via the salvage pathway, although the general increase in most sphingolipids argued against activation of that pathway. Based on the observation that both sphinganine and dihydroceramides were increased (table E2), we hypothesise that hyperoxia activates the pulmonary de novo pathway of ceramide synthesis in the newborn. Further studies focusing on the activity of the rate-limiting enzyme serine palmitoyltransferase would be needed to confirm the increase in the de novo synthesis.
Long chain ceramides have been shown to increase the permeability of endothelium [33], which causes endothelial leakage [34] which might contribute to the reduced compliance of the lungs seen in our hyperoxia model. We postulate that reduced forced expiratory flows might be related to structural changes, such as a reduced number of alveolar attachments, which remained low during air recovery. In contrast, resistance and compliance may be determined by reversible factors in the lung, such as inflammation and endothelial leakage. Our study shows that sphingolipid levels, including ceramides, returned to normal within 4 weeks of recovery in room air, corresponding to the time course of lung function changes. It may well be that when the primary stress factor (hyperoxia) disappears during recovery in room air, this leads to a decrease in sphingolipids and also in lung inflammation and oedema, resulting in partial recovery of lung function. However, no additional effect of d-sphingosine supplementation on partial lung function recovery was observed. Note that sphingolipid levels were measured in BALF, providing a good estimate of the sphingolipid metabolism in the bronchoalveolar compartment, but they may not represent sphingolipid metabolism in the interstitial and vascular compartments. Further research into the complexity of the interaction between these different compartments is warranted.
Treatment with d-sphingosine significantly decreased ceramide levels after 1 and 2 weeks of recovery in room air. Lung morphometry showed an improvement of mean linear intercept and radial alveolar count after 4 weeks of recovery in the d-sphingosine-treated animals compared to untreated control mice. These findings suggest that d-sphingosine supplementation had a beneficial effect on the alveolar injury caused by hyperoxia exposure, and that lower ceramide levels might be responsible for this improvement in alveolar histology. A reduction in the amount of apoptosis is the most likely mechanism by which d-sphingosine acts, but further experiments are needed to confirm this hypothesis.
No obvious beneficial effects on lung function were found, which could well be due to imperfect agreement between measures of lung structure and function. The incomplete recovery of histology, sphingolipid levels and lung function after d-sphingosine treatment and the absence of a significant difference in these parameters between hyperoxic mice with and without d-sphingosine might be due to dose and/or timing of the supplementation, or might have been missed due to relatively small numbers of observations. Our d-sphingosine dose was based on a study by Diab et al. [35] who showed that adult mice injected with d-sphingosine at 20 μg per mouse for five consecutive days had significantly increased sphingosine-1-phosphate (a pro-survival sphingolipid) levels in homogenised lung tissue at day 5 after the start of administration. The administration of d-sphingosine in our model started after hyperoxia exposure and lasted for 5 days. It is possible that earlier or longer might yield better results.
It could be argued that prevention of damage by adding d-sphingosine at an earlier time point would be preferable. We decided to take another approach, as currently there are no reliable predictors to indicate which infant will develop BPD, making it difficult to target preventative treatment to those who might benefit. Therefore, we supplemented d-sphingosine only during the recovery phase to see if this might stimulate repair of hyperoxia-induced damage.
In summary, we demonstrated that hyperoxia in the newborn mouse lung caused a transient increase of sphingolipids, including ceramides, which returned to normal during recovery in room air, while histology remained abnormal. The time course of sphingolipid reduction during recovery matched the normalisation of airway resistance and compliance. Importantly, d-sphingosine supplementation during recovery accelerated the normalisation of ceramides, and significantly improved hyperoxia-induced lung damage. We propose that d-sphingosine and other inhibitors of bioactive sphingolipids should be studied further for their potential to reduce lung damage in human preterm infants who require neonatal oxygen treatment. The ultimate goal would be to develop a new strategy to prevent or correct bronchopulmonary dysplasia.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support statement: This work was supported by an operating grant (MOP-86472) from the Canadian Institutes of Health Research and infrastructure grants (CCURE, CSCCD) from the Canada Foundation for Innovation. Martin Post is the holder of a Canadian Research Chair in Fetal, Neonatal and Maternal Health.
Conflict of interest: None declared.
- Received June 11, 2012.
- Accepted October 23, 2012.
- ©ERS 2013
References










