





Abstract
This study aimed to investigate bronchiolar catalase expression and its relationship with smoking and/or chronic obstructive pulmonary disease (COPD) in humans and to determine the dynamic change of bronchiolar catalase expression in response to cigarette smoke in mice.
Lung tissue was obtained from 36 subjects undergoing surgery for peripheral tumours, consisting of life-long nonsmokers and smokers with or without COPD. Male C57BL/6 mice were subjected to cigarette smoke exposure for up to 3 months followed by a 28-day cessation period. We quantified bronchiolar catalase mRNA using laser capture microdissection and quantitative reverse transcription-polymerase chain reaction. C22 club cells (Clara cells) in culture were exposed to cigarette smoke extract and monitored for viability when catalase expression was decreased by siRNA.
Catalase was decreased at mRNA and protein levels in bronchiolar epithelium in smokers with COPD. In mice, bronchiolar catalase is temporarily upregulated at 1 day after cigarette smoke exposure but is downregulated by repeated cigarette smoke exposure, and is not restored long after withdrawal once emphysema is developed. Decreasing catalase expression in C22 cells resulted in greater cigarette smoke extract-induced cell death.
Bronchiolar catalase reduction is associated with COPD. Regulation of catalase depends on the duration of cigarette smoke exposure, and plays a critical role for protection against cigarette smoke-induced cell damage.
Introduction
Oxidative stress in the lungs has been believed to be a key component of the pathogenesis of chronic obstructive pulmonary disease (COPD) [1]. Animal studies also support an individual ability to defend against cigarette smoke (CS)-induced oxidative stress by upregulation of lung antioxidant defences representing it as a critical event in development of emphysema [2].
Due to their direct contact with the environment, epithelial cells located along the respiratory tract are exposed to CS; therefore, they are likely to be involved in the pathogenesis of smoking-related diseases. In particular, terminal bronchioles are known to play a critical role in a variety of smoking-related lung diseases, and they are the major sites of airflow limitation in COPD [3]. However, there is limited information about bronchiolar epithelial antioxidant defences in smokers, and even less about those in patients with COPD. We have recently investigated the cellular and molecular changes in bronchiolar epithelium in smoking mice [4, 5], as well as in human smokers, and their relationships with the development of COPD [6–8]. In this study, we used laser capture microdissection (LCM) to isolate the terminal bronchiolar epithelium and performed cDNA array analysis, focusing on stress and toxicity pathways, for screening. These data revealed that catalase was an abundantly expressed antioxidant gene in the bronchiolar epithelium of normal (nonsmoker) adult lungs, and furthermore only catalase mRNA was remarkably decreased in the bronchiolar epithelium of patients with COPD.
Catalase, a 240-kDa tetrameric heme protein, plays a central role in the antioxidant screen of the lungs by virtue of its ability to convert hydrogen peroxide to oxygen and water [9]. Catalase is expressed during the later stages of lung development, and is constitutively expressed in airway and alveolar epithelial cells and in macrophages in adults [10]. To date, several studies have focused on catalase in pathological lung status [9–11]. However, no studies have been comprehensively conducted in association of catalase and smoking or COPD. The significance of catalase in pulmonary defence, especially at the bronchiolar level, thus has possibly been underestimated. Unlike most antioxidants, catalase is not elevated in bronchiolar epithelium in healthy human smokers [12].
Accordingly, we have examined the effects of CS exposure on the catalase expression of the cells in the mouse distal airway epithelial cells. Furthermore, we have also investigated whether catalase might play a role in protection against CS-induced damage of immortalised murine club cells (Clara cells) C22.
Materials and methods
Details of the materials and methods used in this study are provided in the online supplementary material.
Collection of human tissue specimens
Lung tissue specimens were obtained from 36 patients undergoing lung resection for small peripheral tumours. COPD patients were chosen based on the guidelines of the Global Initiative for Obstructive Lung Disease [13]. Informed consent was obtained from each subject and the Ethics Committee of Hokkaido University School of Medicine, Sapporo, Japan, approved the study protocols. Some patients had been subjects in our previous study [6–8, 14].
cDNA array analysis of LCM-retrieved bronchiolar epithelial cells
Human bronchiolar epithelial cells were harvested by LCM and total RNA was extracted as described previously [6]. The non-radioactive GEArray Q series cDNA expression array filter containing 96 genes whose expression levels are indicative of stress and toxicity pathways (HS-012N; SuperArray Inc., Bethesda, MD, USA) was applied.
Immunohistochemistry for catalase and semi-quantitative scoring
Immunostaining for catalase in human lungs was performed using rabbit anti-catalase polyclonal antibody (Calbiochem-Novabiochem, San Diego, CA, USA). The catalase immuno-intensity was semi-quantified as described previously by two independent observers in a blind manner, and average scores were reported.
Mouse cigarette smoking models
Male C57BL/6J mice, 9–10 weeks of age (Charles River, Atsugi, Japan), were exposed to whole body mainstream CS for 90 min per day [7] or nose-only mainstream CS for 60 min per day [15]. 3 months of repeated CS exposure using either exposure system results in significant airspace enlargement [7, 15]. Mice were sacrificed and lung samples were collected at several time-points indicated (n=4–6 in each group), and bronchiolar epithelial cells were harvested by LCM as described previously [5].
In situ hybridisation for catalase
Mouse lungs were inflation-fixed with 10% neutral-buffered formalin, paraffin-embedded, and cut into 5-μm sections. Deparaffinised sections were hybridised with a digoxigenin-labelled RNA probe corresponding to nucleotides 1215–1533 of the mouse catalase gene. After hybridisation, digoxigenin detection was performed using the alkaline phosphate-conjugated anti-digoxigenin antibody (Roche, Basel, Switzerland).
Real-time RT-PCR
RNA purification, reverse transcription and quantitative PCR were carried out as described previously [6]. A Taqman Gene Expression Assays probe Hs00156308_m1 was used for human catalase and levels were normalised against glyceraldehyde-3-phosphatase dehydrogenase mRNA, while Mm00437992_m1 was used for mouse catalase and levels were normalised against beta2-microglobulin mRNA (Applied Biosystems, Foster City, CA, USA).
Exposure of CS extract to C22 cells and inhibition of catalase by siRNA
C22 cells were transfected with 20 nM catalase siRNA duplex (Sigma-Aldrich, St. Louis, MO, USA) using INTERFERin siRNA transfection reagent (Polyplus-Transfection Inc., San Marcos, CA, USA) and exposed to diluted CS extract in serum-free media as described previously [4]. In order to assess the cell viability, the cell-free media was assayed for lactate dehydrogenase (LDH) activity as previously described [4].
Data presentation and statistical analysis
All data are expressed as the mean±sem or the median, as appropriate. In humans, differences were analysed using single factor analysis of variance followed by Fisher's protected least significant difference test as a post hoc test or the Kruskall–Wallis test and Mann–Whitney test. In mice, statistical significance was determined by Dunnet multiple comparative analyses.
Results
Characteristics of human subjects
We collected the subjects of three groups: 13 life-long nonsmokers, 13 smokers without COPD and 10 smokers with COPD. Clinical characteristics of the subjects are summarised in table 1. None of the subjects had a history of asthma and none had suffered from acute respiratory infections in the preceding month. Both groups of smokers were of similar pack-years of smoking with various durations of smoking and cessation (table 2). All of the COPD patients exhibited forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) lower than the lower limit of normal [16].
cDNA array for stress and toxicity pathway genes reveals the decrease of catalase in smokers with COPD
To address the question whether altered expression of genes related to stress and toxicity pathways in bronchiolar epithelium might be linked to the chronic smoking histories and/or COPD in humans, cDNA array analysis was performed. Because of limitations in retrievable cells by LCM, it was necessary to pool the cells from subjects in each group (n=10) in order to obtain sufficient RNA without amplification. Among the group of 22 oxidative and metabolic stress-related genes on the cDNA array, catalase was an antioxidant gene most abundantly expressed in bronchiolar epithelium in normal adult lungs, and only catalase showed a more than 1.8-fold decrease in smokers with COPD when compared with nonsmokers (table S1). This is in line with the findings of Ning et al. [17], showing downregulation of catalase in the lungs of moderate COPD patients compared with COPD at-risk patients.
Quantitative analysis confirmed the significant downregulation of catalase expression in bronchiolar epithelium
To confirm the data on cDNA array showing the downregulation of bronchiolar catalase in COPD patients, the RNA harvested from individual bronchiolar epithelium was subjected to quantitative RT-PCR for catalase (fig. 1), consistent with the present cDNA array results (table S1). Among the smokers, the individual expression levels of bronchiolar catalase were not related to pack-years of smoking (fig. S1).

Catalase expression in bronchiolar epithelium. Human bronchiolar epithelial cells were harvested by laser capture microdissection and catalase mRNA was quantified by RT-PCR. Catalase expression was significantly downregulated in bronchiolar epithelial cells of smokers with chronic obstructive pulmonary disease (COPD) when compared with nonsmokers (median 0.6 versus 1.5; p<0.05), while the difference between smokers with and without COPD did not reach the statistical significance (p=0.072). Medians are indicated by horizontal lines. GAPDH: glyceraldehyde phosphate dehydrogenase.
Bronchiolar catalase protein is diminished in human COPD lungs
To assess the decrease of bronchiolar catalase at the protein level in COPD, immunohistochemistry was performed. Catalase was predominantly localised in the apical parts of airway epithelial cells of nonsmokers (fig. 2a). Catalase was not detected in alveolar tissue even in the nonsmokers (data not shown). This is consistent with findings by Kaarteenaho-Wiik and Kinnula [10], showing immunoreactivity of catalase in bronchiolar epithelium in human lungs. However, despite minimal change in bronchiolar catalase in smokers without COPD, a dramatic decrease in catalase staining in the cells of the terminal bronchioles was detected in smokers with COPD (fig. 2b–d). There are no significant correlations between bronchiolar catalase score on immunohistochemistry and percentage FEV1 or degree of emphysema on computed tomography (CT) scan among the COPD patients (data not shown).

Immunohistochemistry for catalase in bronchiolar epithelium. a) nonsmoker, b) smoker without chronic obstructive pulmonary disease (COPD) (age 73 years, 56 pack-years of smoking), c) smoker with COPD (age 77 years, 40 pack-years of smoking). Positive immunohistochemical staining appears blue–purple. Arrows indicate bronchiolar epithelium, immunostained with anti-human catalase antibody. d) By scoring, catalase was significantly attenuated in bronchiolar epithelium in the smokers with COPD, compared with both nonsmokers and smokers without COPD (mean±se 1.1±0.1 versus 2.4±0.2 and 1.9±0.2, respectively; p<0.05). Original magnification (a–c) ×200 (insets ×800).
Catalase expression is enriched in bronchiolar epithelium and is diminished following CS exposure in mice
We next examined the catalase expression in the lungs of mice and examined the effects of CS exposure on catalase expression. In situ hybridisation revealed a high level of catalase mRNA in the epithelial cells lining the terminal airway of non-CS exposed C57BL/6J control mice (figs 3a and c), similar to that seen in human lungs. In the mice subjected to whole-body CS exposure for 10 days, catalase mRNA was diminished (figs 3b and d). Catalase was not seen in alveolar tissue even in non-CS exposed control mice, which matched the findings in human lungs.

Localisation of catalase mRNA in murine lung: images of lung sections hybridised in situ for catalase mRNA. In a normal lung, catalase mRNA is prominent in bronchiolar epithelium from the junction of the terminal bronchiole and alveolar ducts and proximally along the bronchiole up to 250 μm (a and c). At 10 days, following repeated daily cigarette smoke exposure, catalase mRNA is decreased in the bronchiolar epithelium (b and d). e) Controls using a sense probe demonstrated minimal hybridisation in normal lung. These images are representative of a couple of mice in each group. Arrows indicate bronchiolar epithelium hybridised with a digoxigenin-labelled RNA probe corresponding to nucleotides of the mouse catalase gene. a, b, e) Scale bars=600 μm. c, d) Scale bars=20 μm.
Bimodal regulation of catalase expression in bronchiolar epithelium depending on the duration of CS exposure in mice
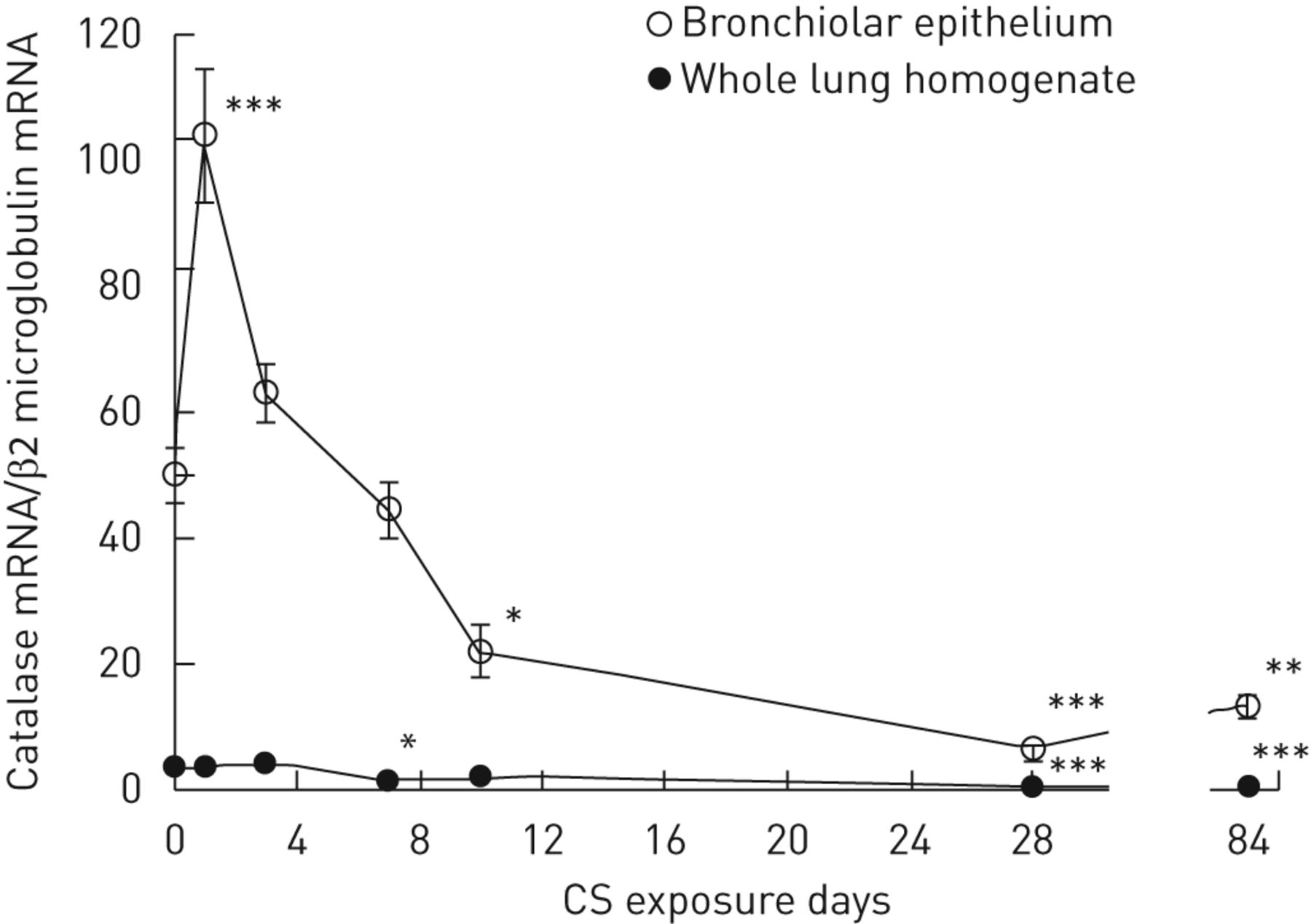
In order to investigate dynamic changes in bronchiolar catalase expression during the development of emphysema, the catalase mRNA expression in LCM-retrieved bronchiolar epithelium and in whole lung tissue was quantified at various time-points following daily whole-body CS exposures up to 3 months. It is noteworthy that catalase mRNA was enriched 14-fold in bronchiolar epithelium when compared with the levels in whole lung tissue in nonexposed adult mice (fig. 4).

Whole-lung and bronchiolar catalase expression in mice during cigarette smoke (CS) exposure. Shown are the time courses for catalase mRNA expression during repeated whole-body CS exposure for 84 days in bronchiolar epithelium, and in whole lung homogenate. Bronchiolar catalase mRNA was temporally upregulated after 1 day of CS exposure compared with non-CS exposed animals (104.1±12.5 versus 50.2±6.0, p<0.001); however, it was downregulated after 10 days (22.1±4.8 versus 50.2±6.0, p<0.05), 28 days (6.5±1.5 versus 50.2±6.0, p<0.001) and 84 days (3 months) of CS exposure (13.4±2.2 versus 50.2±6.0, p<0.01). Catalase mRNA expression in whole lung tissue remained low over time, while it was weakly, but statistically, decreased compared with non-CS exposed mice after 7 days (1.7±0.3 versus 3.5±0.6, p<0.05), 28 days (0.5±0.1 versus 3.5±0.61, p<0.001) and 84 days (3 months) of CS exposure (0.7±0.1 versus 3.5±0.6, p<0.001). Each data point represents the mean±sem for six animals. *: p<0.05, **: p<0.01, and ***: p<0.001 versus non-CS exposed mice.
Similar results were obtained using the nose-only model for CS exposure. Catalase expression in bronchiolar epithelium was decreased after 10 days and 84 days (3 months) in the CS exposed mice, as compared with non-CS exposed mice, along with the increase in CS-induced apoptosis in those cells (fig. 5 and fig. S2). Together, these data suggest that acute CS exposure (1 day) induced an upregulation of catalase in bronchiolar epithelium, while chronic CS exposure (up to 3 months) decreased its expression, coinciding with the development of airspace enlargement as reported elsewhere [7, 15]. This finding is in line with our previous data analysed on microarray [4].

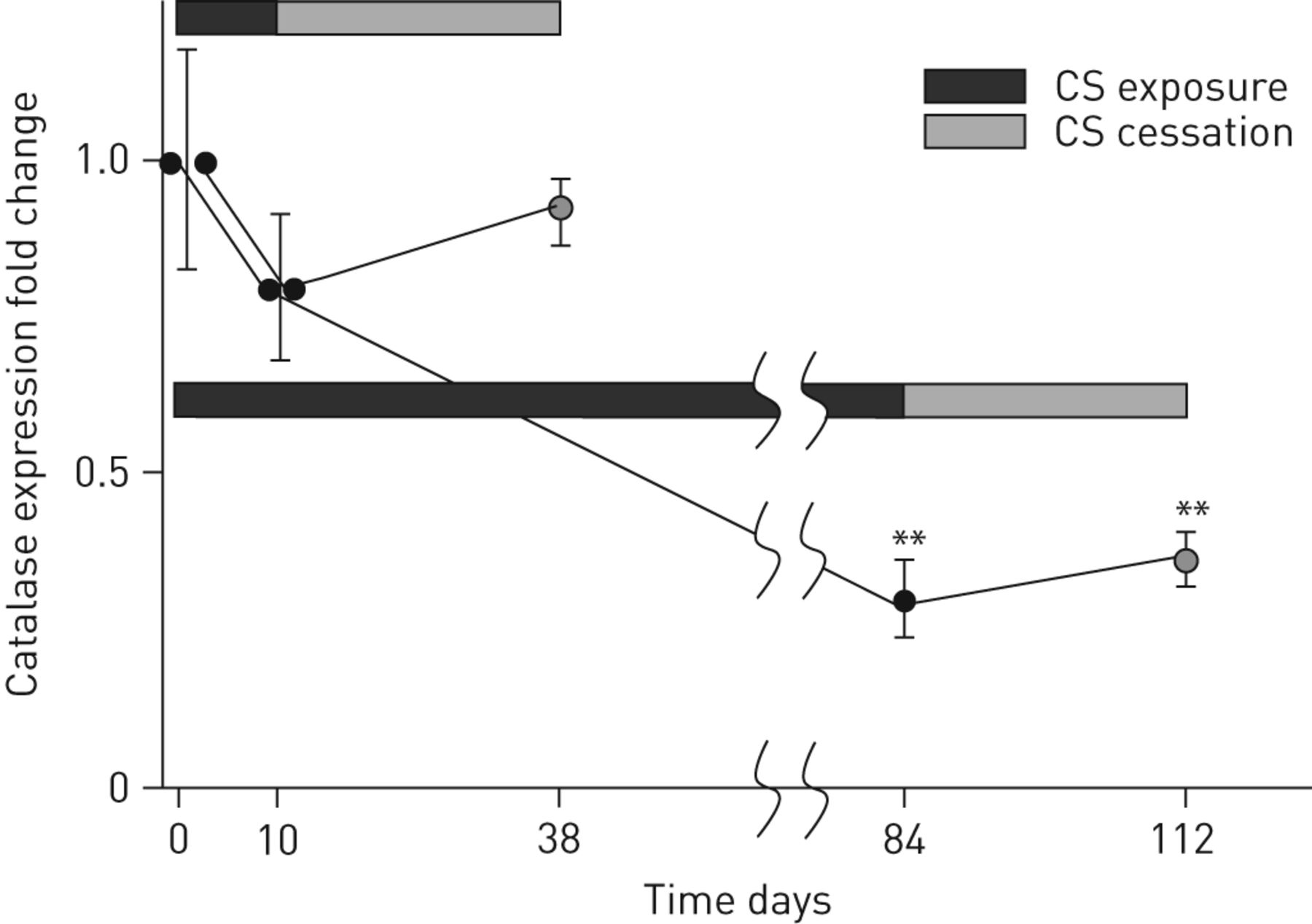
The recovery of bronchiolar catalase expression after the withdrawal of cigarette smoke (CS) exposure. Fold changes of bronchiolar catalase expression after the withdrawal of nose-only CS exposure following short-term (10 days) or long-term (84 days) CS exposure are shown. Each data point represents the mean±sem for 4–6 animals. **: p<0.01 versus non-CS exposed mice.
Downregulated bronchiolar catalase expression is not restored long after discontinuation of chronic smoke exposure in mice
We next addressed the question of whether the withdrawal of CS exposure could restore the bronchiolar catalase expression. As mentioned above, the catalase expression in bronchiolar epithelium after 84 days (3 months) of CS exposure was decreased more than threefold relative to non-CS exposed mice (fig. 5). After 28 days of CS cessation, the catalase expression was slightly elevated; however, the overall level of catalase expression in the bronchiolar epithelium was still suppressed. These are consistent with the findings that impaired expression of catalase persists long after smoking cessation in some of the COPD patients (table 2 and fig. 1).
In contrast, when the 28-day CS cessation period occurred following only 10 days of CS exposure, the bronchiolar catalase expression returned to close to baseline levels (fig. 5). In addition, the duration of CS exposure not only affected the type of inflammatory response to CS (a 4:1 ratio of macrophages to neutrophils after 10 days of CS exposure to 2:1 macrophages to neutrophils plus a fourfold increase in lymphocytes after 3 months of CS exposure), but also the recovery resilience following CS cessation (approximately fivefold versus approximately threefold reduction in total cells after a 28-day cessation period following 10 days or 3 months of CS exposure, respectively (table S2). These data suggest that the ability to recover from the effects of CS exposure (e.g. decrease in catalase expression and inflammation resolution) is more dependent on the duration of CS exposure than the duration of the cessation period.
CS extract induces C22 cell expression of catalase gene
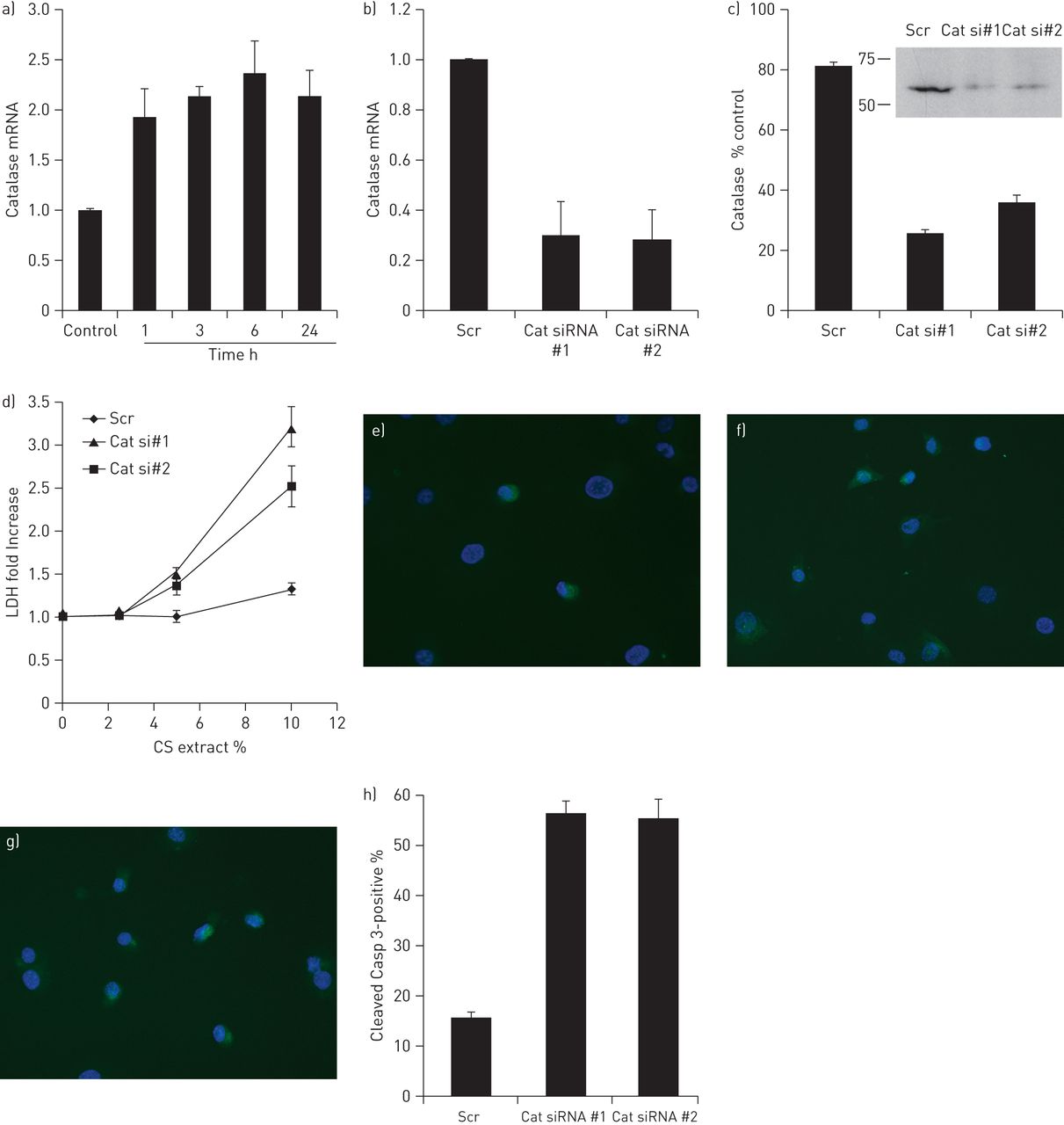
To examine the direct effects of CS on catalase expression in cells lining terminal bronchioles, which are >90% Clara cells in mice, we exposed C22 cells to CS extract in vitro. CS exposure induced a approximately twofold increase of catalase expression in C22 cells at 3, 6 and 24 h compared with the controls (p<0.05) (fig. 6a). This is consistent to the findings that bronchiolar catalase is temporally upregulated at 1 day of CS exposure in vivo in mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Catalase mRNA upregulation in C22 cells in response to cigarette smoke (CS) extract and effects of catalase on CS-induced cell death. a) RNAs isolated from C22 cells exposed to serum-free media containing 10% CS extract for up to 24 h were subjected to real-time RT-PCR for catalase. Following normalisation to β2-microglobulin, results are expressed as fold change above untreated conditions. Data are representative of three independent experiments ±sem in triplicate. *: p<0.05 relative to the control. C22 cells were independently transfected with two different siRNA duplexes targeting catalase expression (Cat), or a scrambled siRNA duplex (Scr). 2 days after transfection, RNAs were isolated and subjected to real-time RT-PCR for expression of catalase (b), and total protein was isolated and subjected to Western blot analysis (c). The C22 cells transfected with catalase siRNAs expressed remarkably decreased levels of catalase mRNA (70% on average) compared with those treated with the scrambled siRNA, which was confirmed at the protein level by Western blotting. In separate experiments, 2 days after transfection with scrambled or catalase siRNA duplexes, C22 cells were exposed to serum-free media containing 10% CS extract for 24 h and the relative amount of LDH in the media was determined. Results are expressed as fold change relative to C22 cells that were transfected with the Scr siRNA exposed to serum-free media alone. In addition, C22 cells that were transfected with Scr siRNA (e), Cat siRNA#1 (f) or Cat siRNA#2 (g) duplexes and exposed to 10% CS extract for 24 h were stained for apoptosis using an anti-cleaved caspase-3 antibody and nuclei were detected with 4',6-diamidino-2-phenylindole. Results are quantified and expressed as the percentage of cleaved caspase-3 positive cells ±sem.
C22 cells with depleted catalase are susceptible to CS-induced cell death
To determine the role of catalase in bronchiolar epithelial cells, C22 cells were transfected with siRNA duplexes for catalase (fig. 6b and c). There are no significant changes in the other antioxidant and detoxification genes when catalase was knocked-down (fig. S3). After 2 days following transfection, cells were exposed to serum-free media either alone or containing various concentrations of CS extract for 24 h, and the level of LDH activity, an indicator of cell death, in the culture media was measured. C22 cells transfected with the scrambled siRNA had slightly increased LDH activity in the culture media when exposed to 10% CS extract as compared with the cells exposed to 0% CS extract. Exposure of C22 cells to ≥20% CS extract resulted in significant cell death, even in C22 cells transfected with the scrambled siRNA [4]. In contrast, cells with decreased catalase expression by siRNA were dramatically more susceptible to CS exposure, as indicated by a significant increase in LDH activity in the conditioned media, even at a concentration of 5% CS extract (fig. 6d). These data suggest that catalase plays a role in the protection against CS-induced bronchiolar epithelial cell death.
Discussion
This study has important findings about catalase in bronchiolar epithelium in humans and mice. In humans, catalase was an antioxidant gene most abundantly expressed in bronchiolar epithelium of adult nonsmokers, whereas bronchiolar epithelial catalase was markedly decreased in the lungs of patients with COPD. The experiments in mice demonstrated that the effects of smoking on bronchiolar catalase expression are time-dependent, increasing early after initial smoke exposure but falling with chronic exposure and remaining low even long after smoke exposure has terminated. We also found that the depletion of catalase in C22 cells increased the susceptibility to CS-induced cell death, implying a critical role of catalase in bronchiolar epithelial cells against CS-induced cell damage.
In the human study, the period of smoking cessation was variable among the smokers. On the one hand, it is possible that the changes in gene expression would have been more substantial if measurements were performed while the subjects were still smoking [18]. On the other hand, this observation particularly acknowledges the fact that not all gene expressions are restored to normal in airway epithelium as a result of giving up smoking. Some nonreversible changes in gene expression, e.g. catalase, might be linked to the progression of the disease even after smoking cessation and/or high risk of development of lung cancer in COPD patients [19]. Having COPD and smoking has a bigger effect than smoking without COPD on the suppression of bronchiolar catalase at the protein level (fig. 2), whereas the statistical difference was not significant between smokers with and without COPD at the mRNA level (fig. 1). This discrepancy suggests that COPD, for unknown reasons, is also associated with a faster turnover or impaired synthesis of catalase protein.
Our time course study in mice indicates that the acute effects of CS exposure cannot be extrapolated confidently to the chronic effects of smoking. The effects of CS exposure on bronchiolar epithelial cells over time may result from several processes having different time frames. Interestingly, the downregulation of bronchiolar catalase persists in mice after the withdrawal of CS exposure once airspace enlargement has developed. These features may mimic the status in former smokers with mild COPD in humans. At transcriptional levels, catalase is directly regulated by FoxO3a and co-activator, peroxisome proliferator-activated receptor γ co-activator 1-α [20]. Hwang et al. [21] recently reported that FoxO3 was predominantly localised in airways/alveolar epithelium in nonsmokers, which was decreased both in lungs of smokers and patients with COPD, and also was decreased in lungs of mice exposed to CS. In that study, the catalase upregulation in mice lungs, in response to CS exposure for 3 days was significantly impaired in FoxO3-deficient mice, although there is no difference in the level of catalase expression in the lungs at steady state between wild-type and Foxo3-deficient mice, suggesting the pivotal role of FoxO3 in transcriptional regulation of catalase. Catalase can also be affected by nuclear factor (NF)-E2-related factor 2 (Nrf2) in responses of the lung to CS [4]. Although the levels of Nrf2 expression were not decreased in bronchiolar epithelial cells [8], genetic or epigenetic inactivation of those transcriptional factors might be involved in the mechanism by which catalase downregulation persists in bronchiolar epithelial cells. These studies emphasise the importance of antioxidant-mediated cell response for protection against CS-induced lung epithelial cell damage. After chronic CS exposure, the oxidative stress becomes greater than the antioxidant potential, along with a downregulation of catalase, and CS-induced apoptosis occurs in bronchiolar epithelial cells.
As catalase handles the intracellular burden of hydrogen peroxide and its toxic derivatives, and hydrogen peroxide reversely inhibits catalase activity and downregulates catalase expression [22], catalase-deficient epithelial cells may release excessively hydrogen peroxide in extracellular milieu. Hydrogen peroxide is not a radical; therefore, it is less reactive and, hence, much more stable than other reactive oxygen species [23]. It has been known that patients with COPD exhale more hydrogen peroxide, regardless of the status of current smoking [24]. Acatalasemic mice are reportedly more susceptible to oxidant tissue injuries, leading to renal fibrosis [25] and peritoneal fibrosis [26]. Hydrogen peroxide scavenging by catalase also suppresses smoke-induced fibrotic remodelling in airways [27]. Therefore, decreased expression of catalase in bronchiolar epithelial cells might be also relevant to peribronchiolar fibrosis in smoking mice [28] and in COPD [3]. Catalase was shown to prevent the redox-sensitive nuclear transcription factor nuclear factor-κB from activating a cascade leading to lung inflammation through rapid regulation of physiologic ROS levels [29]. Catalase prevents apoptosis in human macrophages through the regulation of B-cell lymphoma-2 family protein, implying that the insufficient upregulation and/or suppression of catalase may be involved in apoptosis [30].
CS has been well known to induce cell death of alveolar macrophages, lung endothelial cells, and various lung epithelial cell types. The susceptibility of the lung cells to CS has been shown, at least in part, to be determined by the regulation of protective antioxidant systems. In human studies, besides catalase the other genes of note on the arrays were heat shock 70 kDa protein 1A (HSP70-1A), heat shock 70 kDa protein 1B (HSPA1B), heat shock 70 kDa protein-like 1 (HSPA1L) and heat shock 70 kDa protein 6 (HSP70B), all of which showed >1.5-fold downregulation in smokers with COPD when compared with never-smokers. This is in line with the findings of Ning et al. [17] showing the downregulation of catalase and HSP70s in the lungs of moderate COPD (Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage II) patients when compared with those from COPD at-risk (GOLD 0) patients. Reductions in HSP70s fail to inhibit the activation of NF-κB, which in turn results in enhanced expression of pro-inflammatory genes, such as interleukin-8. This may partly explain how decreases in catalase and HSPs are associated with sustained inflammation in small airways only in susceptible smokers who are developing COPD.
Our study has several limitations. First, although our interests have been focused on the pathogenesis of mild COPD, future investigation of severe COPD patients is crucial for comparison. However, it should be taken into consideration that having even mild airflow limitation and presence of mild parenchymal destructive changes are associated with low levels of catalase in bronchiolar epithelial cells, irrespective of smoking cessation. Secondly, marked differences in sex between the three human groups raise the possibility that sex might be a contributor to the differences in catalase expression in lung cells. However, the bronchiolar catalase expression of smokers with COPD was lower than that of smokers without COPD. Thirdly, there are no measures of catalase activity in this study. However, measuring whole lung catalase activity would completely miss the changes in bronchiolar catalase, as the levels are so low in all lung tissues except for the bronchioles.
We conclude that impaired induction or inactivation of catalase in response to smoke exposure results in unwanted redox imbalance in bronchiolar epithelial cells that may contribute to various smoking-induced lung disorders, including COPD. Our findings suggest bronchiolar epithelium as a cellular target for the development of new anti-oxidant therapeutic approaches, which may protect the lungs from oxidative damage.
Acknowledgments
The authors would like to thank Yoko Suzuki (Hokkaido University School of Medicine, Sapporo, Japan), Jinko Hata (Teijin Pharma Ltd, Tokyo, Japan), Masaru Suzuki (Hokkaido University School of Medicine) for assistance in immunohistochemistry and laser capture microdissection, Gail L. Griffin (Washington University School of Medicine and Barnes-Jewish Hospital, St. Louis, MO, USA) for assistance with ISH, Yasuyuki Nasuhara (Hokkaido University School of Medicine) for scoring computed tomography scan, Ichio Hamamura and Hiroshi Takahashi (both Teijin Pharma Ltd), Katsura Nagai and Takyuki Yoshida (both Hokkaido University School of Medicine) for smoking mice, and Robert M. Senior (Washington University School of Medicine and Barnes-Jewish Hospital) for critical reading of the manuscript.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support statement: This research was supported by the Respiratory Failure Research Group of the Ministry of Health, Labor and Welfare of Japan, Scientific Research Grants from the Ministry of Education, Science, Culture and Sports of Japan, NIH grants P50 HL084922 and PO1 HL29594; and the Alan and Edith Wolff Charitable Trust/Barnes-Jewish Hospital Foundation.
Conflict of interest: None declared.
- Received April 9, 2012.
- Accepted September 21, 2012.
- ©ERS 2013
References










