





Abstract
Cigarette smoking contributes to lung remodelling in chronic obstructive pulmonary disease (COPD). As part of this remodelling, peribronchiolar fibrosis is observed in the small airways of COPD patients and contributes to airway obstruction. Fibroblast-to-myofibroblast transition is a key step in peribronchiolar fibrosis formation.
This in vitro study examined the effect of cigarette smoke on bronchial fibroblast-to-myofibroblast transition, and whether aclidinium bromide inhibits this process. Human bronchial fibroblasts were incubated with aclidinium bromide (10−9–10−7 M) and exposed to cigarette smoke extract. Collagen type I and α-smooth muscle actin (α-SMA) expression were measured by real-time PCR and Western blotting, as myofibroblast markers. Intracellular reactive oxygen species, cyclic AMP (cAMP), extracellular signal-regulated kinase (ERK)1/2 and choline acetyltransferase were measured as intracellular signalling mediators.
Cigarette smoke-induced collagen type I and α-SMA was mediated by the production of reactive oxygen species, the depletion of intracellular cAMP and the increase of ERK1/2 phosphorylation and choline acetyltransferase. These effects could be reversed by treatment with the anticholinergic aclidinium bromide, by silencing the mRNA of muscarinic receptors M1, M2 or M3, or by the depletion of extracellular acetylcholine by treatment with acetylcholinesterase.
A non-neuronal cholinergic system is implicated in cigarette smoke-induced bronchial fibroblast-to-myofibroblast transition, which is inhibited by aclidinium bromide.
Chronic obstructive pulmonary disease (COPD) is characterised by airflow limitation that is progressive and not fully reversible. Cigarette smoking is the main risk factor for COPD and contributes to structural changes in airways during COPD progression [1]. Structural changes in COPD patients are characterised by loss of alveolar wall (emphysema), vascular remodelling with pulmonary hypertension, mucus hypersecretion or peribronchiolar fibrosis [1]. As part of fibrotic alterations in COPD, structural changes are seen primarily in small airways. The severity of the disease appears to correlate with thickening of the walls of small airways caused by fibrosis and infiltration of inflammatory cells, which contributes to airflow obstruction [2]. Accumulation and persistence of myofibroblasts is believed to contribute to the development of small airway fibrosis. In this respect, under chronic inflammatory conditions, resident lung fibroblasts are activated and transformed into a more contractile, proliferative and secretory-active myofibroblast phenotype, characterised by increased expression of extracellular matrix components and α-smooth muscle actin (α-SMA), which contributes to the increase of lung remodelling progression and airway bronchoconstrictor responsiveness [3].
Parasympathetic activity is increased in the airways of COPD patients and is the basis for the use of anti-cholinergic therapy [4]. Anti-cholinergics constitute a particularly important bronchodilator therapy in COPD and certain forms of asthma [5]. Furthermore, in animal models, anticholinergics have shown potential anti-inflammatory and antiremodelling effects [6], which may be of additional value to their classical bronchodilator effects.
Recently, it has been proposed that acetylcholine in the airways may be released by non-neuronal cell types, such as airway epithelial cells and lung fibroblasts. Therefore, a dysfunction of a non-neuronal cholinergic system may contribute to the pathophysiology of asthma and COPD [7]. It has been shown that anticholinergic treatment inhibits cigarette smoke-induced mucin hypersecretion in human bronchial epithelial cells [8], as well as cigarette smoke-induced lung fibroblast proliferation [9]. Furthermore, choline acetyltransferase (ChAT), the intracellular enzyme responsible for acetylcholine production, is upregulated in both lung fibroblasts from COPD patients and fibroblasts stimulated with cigarette smoke [9]. However, no data exist concerning the role of cigarette smoke on fibroblast to myofibroblast transition, as well as the potential inhibitory effect of anticholinergics.
Aclidinium bromide is a novel, inhaled, long-acting muscarinic antagonist compound with low systemic activity that has completed phase III clinical development for COPD treatment [10]. In preclinical studies, aclidinium bromide demonstrated potent muscarinic-antagonist activity, comparable to that of ipratropium bromide and tiotropium bromide, and a long duration of action [11]. In clinical trials, aclidinium bromide has provided sustained bronchodilation, similar to that observed with tiotropium bromide, a good safety profile and low incidence of anticholinergic adverse events [10].
In this study, we investigated the role of aclidinium bromide on the increase of the myofibroblast markers collagen type I and α-SMA elicited by chronic cigarette smoke exposure in human bronchial fibroblasts, as well as the intracellular pathways involved in this process.
METHODS
See the online supplementary material for further details of the methods used.
Isolation and cultivation of human fibroblasts
Human bronchial fibroblasts were obtained from patients undergoing surgery for lung carcinoma who gave informed consent, as previously described [12]. Cultivation and characterization of fibroblasts were performed as described elsewhere [12]. The protocol for obtaining human tissue was approved by the local ethical review board for human studies (General Hospital of Valencia, Valencia, Spain). See the online supplementary material for details.
Preparation of cigarette smoke extract and incubations
Cigarette smoke extract (CSE) solutions were prepared as previously described [13]. Briefly, the smoke of a research cigarette (2R4F; Tobacco Health Research, University of Kentucky, Lexington, KY, USA) was bubbled into a flask containing 25 mL pre-warmed (37°C) Dulbecco’s Modified Eagle’s Medium. The resulting solution was defined as CSE at 100%. CSE at 10% reportedly corresponds to the exposure associated with smoking approximately 1–2 packs per day [14]. Before stimulation, subconfluent cell monolayers were deprived of serum for 24 h. Human bronchial fibroblasts were stimulated with CSE (0–10%) for different periods of time (0–72 h), replacing the culture medium and stimulus every 24 h. Different drug modulators were added 30 min before the stimulus. See the online supplementary material for details.
Real-time RT-PCR
Total RNA was isolated from cultured human bronchial fibroblasts by using TriPure® (Roche, Indianapolis, IN, USA), and reverse-transcribed and amplified with specific primers. Relative quantification of these different transcripts was determined with the 2-ΔΔCt method using glyceraldehyde phosphate dehydrogenase as an endogenous control (Applied Biosystems, Foster City, CA, USA) and normalised to the control group. See the online supplementary material for details.
Transfection of small interfering RNAs
Small interfering RNA (siRNA) for M1, M2 and M3 receptors and the scrambled siRNA control were purchased from Ambion (Cambridge, UK). The transfection reagent used was Lipofectamine 2000 (Invitrogen, Paisley, UK) at a final concentration of 2 μL·mL−1. See the online supplementary material for details.
Western blotting
Western blot analysis was used to detect changes in collagen type I (138 kDa), α-SMA, phosphorylate extracellular signal-regulated kinase (ERK)1/2 (42−44 kDa), M1 (52 kDa), M2 (70 kDa), M3 (75 kDa), p67phox (67 kDa), NADPH oxidase (NOX)4 (67 kDa) and ChAT (65 kDa) in bronchial fibroblast lysates. See the online supplementary materials for details of Western blot analyses and antibodies used.
Dichlorodihydrofluorescein fluorescence measurement of reactive oxygen species
2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA; Molecular Probes, Nottingham UK) was used to monitor the intracellular reactive oxygen species (ROS) in bronchial fibroblasts. See the online supplementary material for details.
Cyclic AMP assay
Human lung fibroblasts were cultured in 96-well plates to ∼95% confluence. Following different treatments, cells were lysed and intracellular cyclic AMP (cAMP) content was determined with the cAMP Biotrak enzyme immunoassay according to manufacturer’s instructions (Amersham, Cambridge, UK). Results were expressed as femtomoles per well.
Analysis of results
Data presented as mean±sem of n experiments. Statistical analysis of data was performed by ANOVA followed by the Bonferroni test (GraphPad Software Inc., San Diego, CA, USA). Significance was accepted when p-values were <0.05.
RESULTS
Aclidinium bromide attenuates CSE-induced myofibroblast markers in human bronchial fibroblast cultures
CSE, at 2.5% concentration, upregulated collagen type I and α-SMA in a time-dependent manner, reaching peak values following 48 h of CSE 2.5% exposure (fig. 1a and b). Furthermore, CSE dose-dependently increased collagen type I and α-SMA mRNA and protein expression, reaching statistical significance at 2.5% concentration following 48 h of exposure (fig. 1c–f). Thus, we selected this CSE concentration to evaluate myofibroblast marker expression in future experiments.

Cigarette smoke extract (CSE) time and dose-dependently increased collagen type I and α-smooth muscle actin (α-SMA) in human bronchial fibroblasts. Human lung fibroblasts were stimulated with CSE a, b) at the indicated times or c–f) for 48 h at the indicated concentrations. a–d) After incubation, the RNA was extracted and subjected to RT-PCR with collagen type I- and α-SMA-specific primers and probes. e, f) After incubation, total protein was extracted and Western blots were performed with specific antibodies for collagen type I and α-SMA. e, f) Data represent densitometries of three different Western blots. Each bar represents the mean±sem of a–d) four or e, f) three independent experiments. a–f) One-way repeated-measures ANOVA p<0.001. *: p<0.05 with post hoc Bonferroni test compared with solvent controls.
In other experiments, aclidinium bromide was added 30 min before CSE 2.5% and further incubated for 48 h. Aclidinium bromide dose-dependently reduced the CSE-induced collagen type I and α-SMA mRNA and protein expression, reaching a maximal inhibitory value at 10−7 M (fig. 2a–c). Similar results were observed for atropine, reaching significant inhibition of both myofibroblast markers at 1 μM, suggesting the participation of a cholinergic pathway in this process (fig. 2d–f). Neither aclidinium nor atropine by themselves showed any effect on myofibroblast markers (figs S1 and S2).

Aclidinium bromide (ACL) dose-dependently reduced the cigarette smoke extract (CSE)-induced collagen type I and α-smooth muscle actin (α-SMA) in human bronchial fibroblasts. Human lung fibroblasts were stimulated with CSE for 48 h. ACL or atropine (ATR) were added to the medium at the indicated concentrations 30 min before CSE. a, b, d, e) Total RNA and c, f) protein were extracted after the incubation period. RT-PCR for a, d) collagen type I or b, e) α-SMA. c, f) Densitometry of collagen type I or α-SMA protein expression relative to β-actin and normalised to solvent controls. Representative Western blots of collagen type I and α-SMA are shown. Each graph represents the mean±sem of three experiments for Western blots and four independent experiments for RNA experiments. a–c) One-way repeated-measures ANOVA p<0.001. *: p<0.05 with post hoc Bonferroni test compared with solvent controls; #: p<0.05 with post hoc Bonferroni test compared with stimulus.
Aclidinium bromide reduces intracellular ROS elevated by CSE in human bronchial fibroblasts
In our experiments on human primary lung fibroblasts, CSE (2.5–10%) dose-dependently increased intracellular ROS generation, reaching a significant value after 2 h of stimulation that was sustained for 24 h (fig. 3a). Pretreatment of bronchial fibroblasts with aclidinium bromide dose-dependently reduced the CSE-induced ROS by nearly 50% at 10−7 M after 24 h of CSE 2.5% stimulation (fig. 3b–e). In parallel experiments, quenching of ROS by apocynin (100 μM) or N-acetyl-l-cysteine (NAC) (1 mM), as well as increasing cAMP with its analogue dibutyryl-cAMP (dbcAMP), suppressed CSE 2.5%-triggered intracellular ROS production (fig. 3e).
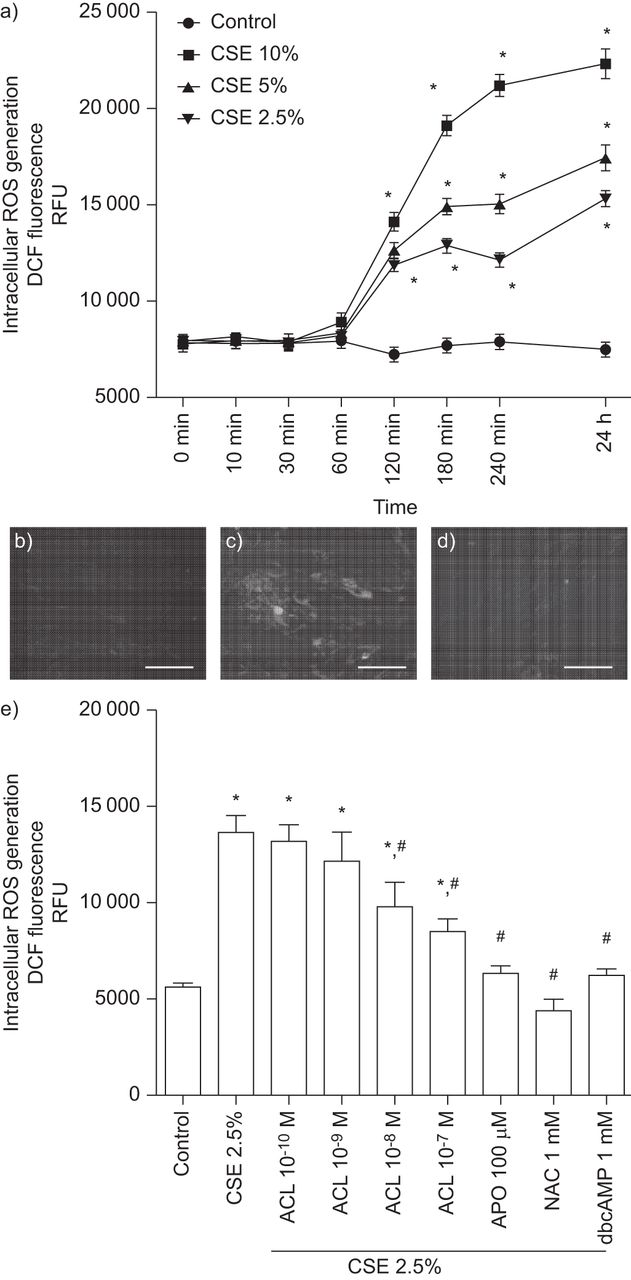
Aclidinium bromide (ACL) mitigated cigarette smoke extract (CSE)-induced intracellular reactive oxygen species (ROS) generation in human bronchial fibroblasts. a) Human bronchial fibroblasts were loaded with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) 30 min before CSE stimulation. CSE dose- and time-dependently increased intracellular ROS in human bronchial fibroblasts. b–e) Human bronchial fibroblasts were loaded with H2DCF-DA in the presence or absence of ACL (10−10–10−7 M), apocynin (APO; 100 μM), N-acetyl-l-cysteine (NAC; 1 mM), dibutyryl cyclic AMP (dbcAMP; 1 mM) or vehicle for 30 min. Excess H2DCF-DA was removed by washing with PBS. ACL, APO, NAC, dbcAMP or vehicle were appropriately replenished before cells were exposed to CSE. b–d) Representative dichlorodihydrofluorescein (DCF) fluorescence images following 24 h of basal or CSE 2.5% stimulation in presence or absence of ACL: b) control; c) CSE; d) 10−7 M ACL plus CSE. Scale bars=25 μm. e) ACL dose-dependently attenuated the CSE 2.5%-induced intracellular ROS after 24 h of stimulation. Similar results were found for APO, NAC and dbcAMP. Results represent the mean±sem of three independent experiments. RFU: relative fluorescence units. a, e) One-way repeated-measures ANOVA p<0.001. *: p<0.05 with post hoc Bonferroni test compared with solvent controls; #: p<0.05 with post hoc Bonferroni test compared with stimulus.
The NADPH oxidase complex is comprised of several cytosolic and plasma membrane subunits [15]. Among the subunits analysed, we found that the cytosolic subunit p67phox and the plasma membrane subunit NOX4 were the most highly expressed in bronchial fibroblasts under basal conditions (fig. 4a). Following 24-h exposure, CSE 2.5% upregulated both mRNA transcripts and protein expression of p67phox and NOX4, which were dose-dependently reverted to near control values by exposure to aclidinium bromide 10−7 M (fig. 4b–d).

Aclidinium bromide (ACL) attenuated the cigarette smoke extract (CSE)-induced p67phox and NADPH oxidase (NOX)4 upregulation in human bronchial fibroblasts. a) RNA was obtained from primary human bronchial fibroblasts and mRNA transcripts for NOX1, p67phox, p47phox and NOX4 were quantified and normalised, setting p67phox as 100%. ACL dose-dependently attenuated the CSE 2.5%-induced p67phox and NOX4 b, c) mRNA and d) protein upregulation following 24 h of stimulation. Results represent the mean±sem of three independent experiments. a–c) One-way repeated-measures ANOVA p<0.001. *: p<0.05 with post hoc Bonferroni test compared with solvent controls; #: p<0.05 with post hoc Bonferroni test compared with stimulus.
CSE activation of non-neuronal cholinergic system is inhibited by aclidinium bromide
Previous reports have indicated that CSE activates a non-neuronal cholinergic system in lung fibroblasts, but the mechanism remains unclear. Since ChAT is the intracellular enzyme responsible for acetylcholine synthesis, we next explored the effect of CSE on ChAT expression. In this regard, CSE 2.5% increased ChAT expression, which was dose-dependently inhibited by aclidinium bromide and by the antioxidants apocynin 100 μM and NAC 1 mM (fig. 5a).

Cigarette smoke extract (CSE) activated a non-neuronal cholinergic system, decreased cyclic AMP (cAMP) and phosphorylated extracellular signal-regulated kinase (ERK)1/2. Human bronchial fibroblasts were pre-incubated for 30 min with a–c) aclidinium bromide (ACL; 10−9–10−7 M), a, b and d) apocynin (APO; 100 μM), N-acetyl-l-cysteine (NAC; 1 mM) or b, d) acetylcholinesterase (AChE; 10 U·mL−1) followed by the stimulation with CSE at 2.5% over 24 h. a) Total protein was extracted and Western blots for choline acetyltransferase (ChAT) and β-actin, as an internal control, were performed. Representative Western blots are shown from three different experiments. b) Cells were lysed and intracellular cAMP was quantified using the cAMP Biotrak enzyme immunoassay system (Amersham, Cambridge, UK). c, d) Total protein was extracted and phosphorylated ERK1/2 (pERK1/2) and total ERK1/2 (as an internal control) protein expression were determined by Western blots. a, c, d) Densitometries of three different Western blots. Results represent the means±sem of three to four independent experiments. One-way repeated-measures ANOVA p<0.01. *: p<0.05 with post hoc Bonferroni test compared with solvent controls; #: p<0.05 with post hoc Bonferroni test compared with stimulus.
Intracellular levels of cAMP and phosphorylation of ERK1/2 have been related to the activation of human lung fibroblasts [12, 16]. In this work, we observed that CSE 2.5% reduced intracellular cAMP levels and increased the ERK1/2 phosphorylation following 24 h of exposure (fig. 5b–d). Aclidinium bromide pretreatment dose-dependently reduced the cAMP downregulation as well as reducing the increase of ERK1/2 phosphorylation (fig. 5b and c). Furthermore, the antioxidant treatment with apocynin 100 μM or NAC 1 mM also reversed the CSE-induced cAMP downregulation and ERK1/2 phosphorylation (fig. 5b and d). Since CSE may activate a non-neuronal cholinergic system, we added the enzyme acetylcholinesterase (AChE) (10 U·mL+1) to remove any extracellular acetylcholine during the 24-h period of CSE 2.5% stimulation. AChE partially reversed cAMP downregulation and ERK1/2 phosphorylation induced by CSE (fig. 5b and d).
The antioxidants NAC 1 mM and apocynin 100 μM, as well as the cAMP analogue dbcAMP and the ERK1/2 inhibitor PD98059 10 μM, partially suppressed the CSE-induced collagen type I and α-SMA mRNA and protein expression (fig. 6a and b). Moreover, both aclidinium bromide 10−7 M and AChE 10 U·mL−1 were also able to suppress the CSE-induced collagen type I and α-SMA mRNA and protein expression in human bronchial fibroblasts, which implicates a non-neuronal cholinergic system in the upregulation of myofibroblast markers (fig. 6a and b). To further study the role of this non-neuronal cholinergic system in the myofibroblast transformation, lung fibroblasts were incubated with the inhibitor of choline uptake transporter hemicholinium-3 at 50 μM or with the AChE inhibitor neostigmine 10 μM before CSE 2.5% exposure. Hemicholinium-3 significantly inhibited the CSE-induced collagen type I and α-SMA mRNA and protein expression, while neostigmine significantly increased the CSE-induced collagen type I and α-SMA mRNA and protein expression (fig. 6c and d). In the absence of CSE, the different drugs assayed did not show any effect on mRNA or protein expression of collagen type I and α-SMA (figs S3 and S4).

The cigarette smoke extract (CSE)-induced collagen type I and α-smooth muscle actin (α-SMA) are partially mediated by a non-neuronal cholinergic pathway. Human bronchial fibroblasts were pre-incubated for 30 min with aclidinium bromide (ACL; 10−7 M), apocynin (APO; 100 μM), N-acetyl-l-cysteine (NAC; 1 mM), PD98059 (10 μM), dibutyryl cyclic AMP (dbcAMP; 1 mM), acetylcholinesterase (AChE; 10 U·mL−1), hemicholinium (HC)-3 (50 μM) or neostigmine at 10 μM followed by the stimulation with CSE at 2.5% over 24 h. a, c) Total RNA or b, d) protein were extracted to quantify mRNA transcripts and protein expression of collagen type I and α-SMA using appropriate primers and antibodies. a, c) Values of collagen type I and α-SMA mRNA expression normalised to control group using glyceraldehyde phosphate dehydrogenase as an internal control. b, d) Densitometry of collagen type I or α-SMA protein expression relative to β-actin and normalised to solvent controls. Representative Western blots of collagen type I and α-SMA are shown. Each graph represents the mean±sem of three to four independent experiments for Western blots and four for RNA experiments. a, b) One-way repeated-measures ANOVA p<0.05. *: p<0.05 with post hoc Bonferroni test compared with solvent controls; #: p<0.05 with post hoc Bonferroni test compared with stimulus.
CSE upregulation of myofibroblast markers is partially mediated by M1, M2 and M3 receptors
Human bronchial fibroblasts transfected with siRNA for M1, M2 or M3 selectively suppressed specific muscarinic receptors without affecting other muscarinic receptors (fig. 7a and b). In this line, siRNA for all three muscarinic receptors showed suppression of CSE-induced myofibroblast markers collagen type I and α-SMA (fig. 7c), indicating that all three muscarinic receptors may be involved in mediating the induction of the myofibroblast markers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cigarette smoke extract (CSE) increases myofibroblast markers by a non-neuronal cholinergic signalling mediated by muscarinic (M)1, M2 and M3 receptors. Human bronchial fibroblasts were selectively transfected with small interfering RNA (siRNA; 50 nM) for M1, M2 and M3 or with the negative control siRNA (-) for 48 h using Lipofectamine 2000 (Invitrogen, Paisley, UK) at final concentration of 2 μg·mL−1. a, b) All siRNA targeting M1, M2 and M3 genes were able to reduce mRNA and protein expression for M1, M2 and M3 significantly without effecting other M receptors. β-actin was assayed as a positive control. Cells transfected with siRNA for M1, M2 and M3 genes attenuated the CSE 2.5%-induced collagen type I and α-smooth muscle actin (α-SMA) protein upregulation. Results are the mean±sem of three independent experiments and are representative of three independent experiments per condition for Western blots. a) One-way repeated-measure ANOVA p<0.001. *: p<0.05 with post hoc Bonferroni test compared with siRNA (-) controls.
DISCUSSION
The main and novel results of this study are that: 1) CSE increased the myofibroblast markers collagen type I and α-SMA in human bronchial fibroblast through a mechanism mediated by increase of intracellular ROS, depletion of cAMP and phosphorylation of ERK1/2; 2) CSE activated a non-neuronal cholinergic system by means of overexpression of ChAT, mediating increase of myofibroblast markers; and 3) the anticholinergic aclidinium bromide was able to attenuate the CSE-induced myofibroblast markers through inhibition of ROS generation, cAMP depletion, ERK1/2 phosphorylation and ChAT overexpression induced by CSE. These new findings provide in vitro evidence of the antiremodelling effect of aclidinium bromide on human bronchial fibroblasts in those cigarette smoke exposure situations that may contribute to the amelioration of the peribronchiolar fibrosis observed in smokers with COPD.
It is known that airflow limitation in COPD patients occurs in distal airways considered as noncartilaginous conducting airways with an internal diameter <2 mm. In this work, we carefully dissected small bronchi of ∼2 mm to obtain bronchial fibroblasts, which may represent the place where peribronchiolar fibrosis occurs [17]. Bronchiolar fibroblast transformation into myofibroblasts is considered a key step in the process of increasing the thickness of the small airways, reducing airways radius and enhancing airflow limitation. Myofibroblasts share phenotypic characteristics with fibroblasts and airway smooth muscle cells. In this regard, myofibroblasts are characterised by the secretion of extracellular matrix components (e.g. collagen type I), a characteristic that is shared with fibroblasts but not with smooth muscle cells, and by formation of contractile apparatus (e.g. α-SMA), a characteristic that is shared with airway smooth muscle cells but not with fibroblasts [3]. Therefore, we selected both of these molecular markers to analyse the myofibroblast-like phenotype. Several growth factors and pro-inflammatory mediators, such as transforming growth factor (TGF)-β1, interleukin-13 and connective tissue growth factor, have been described as inducers of myofibroblast transition [18]; however, no data were available on cigarette smoke, the main risk factor for COPD. In this work, we prepared CSE as we and others have described previously [13, 14], which approximately corresponds to exposures associated with smoking slightly fewer than 0.5 packs per day to slightly fewer than 2 packs per day of cigarettes for CSE 2.5% and 10%, respectively. Currently, several in vitro studies have been focused on effect of CSE on lung fibroblasts; however, no data are available on effect of CSE on bronchial fibroblast to myofibroblast transition. In this work, we observed that CSE promoted myofibroblast marker overexpression, which is compatible with the myofibroblast-like phenotype [3]. Furthermore, the CSE-induced expression of myofibroblast markers was mediated by intracellular ROS production, which was significantly increased after 2 h of exposure and persisted until at least 24 h. The ROS increase preceded the collagen type I and α-SMA overexpression (after 48 h), suggesting a role of intracellular ROS as second messenger. A similar time-response of ROS production has previously been reported in human primary lung fibroblasts [19]. Intracellular ROS production in response to CSE is mediated by direct activation of the NADPH oxidase complex [20]. The NADPH oxidase complex is comprised several cytosolic and plasma membrane units, which vary depending on the cell type. Thus, in human lung fibroblasts, the NADPH oxidase components p47phox, p67phox, p22phox and NOX4 have been observed [21]. In this work, we found that the NADPH oxidase subunits p67phox and NOX4 were the most highly expressed in bronchial fibroblasts. The cytosolic p67phox subunit is mobilised under certain conditions to activate the plasma membrane plasmatic subunits NOX1, NOX2 or NOX3 to produce superoxide and/or hydrogen peroxide. In contrast, the plasma membrane unit NOX4 does not require interaction and activation by cytosolic regulatory subunits, so its activation is directly related to its expression [15]. In this regard, we found that CSE induced the upregulation of both p67phox and NOX4 after 24 h of stimulation, which was in accordance with the increase of intracellular ROS.
Recent reports have related expression of NOX4 as a key factor of fibroblast progression and fibroblast-to-myofibroblast transition [22]. Therefore, a treatment diminishing NOX4 and, in turn, intracellular ROS, may prevent myofibroblast transition. We found that 24-h exposure to either the antioxidant NAC or the NADPH oxidase inhibitor apocynin inhibited the CSE-induced p67phox and NOX4 upregulation. These antioxidant modulators were also able to reduce myofibroblast markers induced by CSE, establishing a link between oxidative stress and myofibroblast transition. The anticholinergic aclidinium bromide attenuated myofibroblast markers induced by CSE and this action was mediated in part by the inhibition of the p67phox and NOX4 expression, as well as by the consequent reduction of intracellular ROS generated by CSE. These results suggest that CSE activates a non-neuronal cholinergic system. Previous reports have suggested that cigarette smoke may activate a non-neuronal cholinergic system in different cell types, including airways and human lung fibroblasts. For example, we have recently shown that CSE promotes synthesis and release of the mucin MUC5AC in differentiated bronchial epithelial cells by a mechanism mediated by release of acetylcholine, and can be inhibited by aclidinium bromide [8]. Furthermore, human lung fibroblasts from COPD patients have shown an increase of muscarinic receptors, as well as ChAT expression, an effect that was mimicked in healthy lung fibroblasts after CSE exposure [9]. In our experiments, AChE prevented the CSE-induced expression of myofibroblast markers, which is consistent with the presence of a non-neuronal cholinergic system. Other evidence to support the activation of a non-neuronal cholinergic system by cigarette smoke includes the upregulation of ChAT, the intracellular enzyme responsible of acetylcholine synthesis. We observed that CSE upregulated ChAT expression, which was prevented by the antioxidants NAC and apocynin, as well as by aclidinium bromide, suggesting the participation of intracellular ROS. Further evidence of the participation of non-neuronal cholinergic system was supported by the inhibition of myofibroblast transition blocking choline uptake with hemicholinium and increasing myofibroblast markers inhibiting AChE with neostigmine.
Nowadays, it is believed that cigarette smoke contains >6,000 compounds and, possibly, this list will grow in line with the new analytical techniques available [23]. Taking into account this assertion, we know that CSE has the advantage of containing all of the compounds inhaled by smokers. However, due to the very complexity of CSE, it is difficult to identify the specific agent mediating a precise effect because of differences found for particular concentrations and durations of exposure of a given agent of CSE. To that end, one can perform a dose–response and time-course analysis and make some crude calculations to suggest that exposure mimics what might happen in vivo. Thus, for example, low concentrations of CSE (<5%) have shown proliferative effects on lung fibroblasts [24–26], while higher (>10%) concentrations showed inhibitory effects on lung fibroblast proliferation [27], thus indicating that among the ∼6,000 substance of cigarette smoke, there are some proliferative and other anti-proliferative compounds that may act depending on their final balance.
In the present work, we have used 2.5% CSE, which is in agreement with the low doses of CSE demonstrating proliferative effects in previous studies [24–26], which support fibroblast activation in the process of myofibroblast transition [28].
Preliminary data from our laboratory indicated that CSE at 2.5% concentration had a slight increase of fibroblast proliferation after 48 h of exposure (based on a bromodeoxyuridine incorporation assay; data not shown). However, this increase was not enough to perform inhibitory experiments with aclidinium bromide. This was the main reason to discard these experiments, since the well-established myofibroblast markers collagen type I and α-SMA are more reliable in mechanistic studies [3].
However, even if one could calculate exposure, experiments performed with CSE cannot mimic all of the components of the microenvironment that exist in living systems (e.g. cell–cell interactions). Thus, despite limitations, results from these studies allow determination of the capacity of CSE to influence cellular functions while eliminating other variables.
Intracellular cAMP is a second messenger that mediates a high number of anti-inflammatory processes. In addition to its anti-inflammatory actions, cAMP also controls the inhibition of fibroblast activation, as well as the myofibroblast transition [29]. Thus, a decrease of the levels of cAMP could promote fibroblast to myofibroblast transition. This is evidenced by TGF-β1, which increases the expression and activity of phosphodiesterases, the enzymes that degrade cAMP, promoting myofibroblast transition [30]. In the case of cigarette smoke, it has been shown that hydrogen peroxide, a component of tobacco smoke, swiftly elevates the activity of phosphodiesterase 4D3 attributed to phosphoinositide 3-kinase and ERK-dependent phosphorylation [31]. In this respect, we have previously observed that CSE reduces intracellular cAMP levels in human differentiated bronchial epithelial cells by means of phosphodiesterase 4B upregulation (data not shown). In this work, we observed that CSE decreases intracellular levels of cAMP and that this effect was partially reversed by the antioxidant NAC and aclidinium bromide, suggesting a role of intracellular ROS. Furthermore, AChE also attenuated the CSE-reduced cAMP, which indicates the involvement of the non-neuronal cholinergic system. This may be explained by the fact that the main muscarinic receptor expressed on lung fibroblasts is M2 [32], which is coupled to Gi protein, so stimulation of M2 by means of a non-neuronal cholinergic system could also promote the inhibition of adenylate cyclase/cAMP pathway and, therefore, myofibroblast transition.
In line with our results, recently, it has been found that prostaglandin (PG)E2, as a potent cAMP enhancer, may inhibit the activation of fibroblasts and their transformation into myofibroblasts [33, 34]. Thus, we cannot discount that one of the mechanisms of aclidinium bromide mediating the increase of cAMP and, consequently, the inhibition of myofibroblast transition could be the increase of PGE2 release.
Another pathway we investigated was ERK1/2 signalling. Previous studies have shown that ERK1/2 participates in fibroblast to myofibroblast transition [35] and that CSE directly phosphorylates ERK1/2 [9]. Thus, CSE-induced myofibroblast transition could be mediated in part by ERK1/2. In this work, we observed that CSE induced the phosphorylation of ERK1/2, which was reduced by aclidinium bromide and by the antioxidant treatment. Moreover, AChE also attenuates the CSE-induced ERK1/2 phosphorylation, suggesting that the activation of muscarinic receptors is also implicated. Previous results support the notion that muscarinic receptor activation increases collagen expression in human lung fibroblasts by means of ERK1/2 activation [12]. Therefore, a non-neuronal cholinergic system may also be participating in the process of CSE-induced ERK1/2 phosphorylation. Thus, the role of ERK1/2 in the CSE-induced myofibroblast markers was confirmed by the inhibitory effect of the ERK1/2 antagonist PD98059.
Based on these results, we may conclude that CSE activates muscarinic receptors in human bronchial fibroblasts by a non-neuronal cholinergic system, and that this mechanism is involved in the upregulation of the myofibroblasts markers. However, which isoform of muscarinic receptor plays a significant role in this mechanism is unclear. It has been shown that the inhibition of the Gi protein with pertussis toxin reduces the fibroblast proliferation and collagen expression observed in response to muscarinic agonists [12], suggesting a key role for the M2 receptor. However, antagonists for M1, M2 and M3 were all effective at inhibiting the fibroblast proliferation induced by acetylcholine [9], although the specific muscarinic antagonists available are not fully selective [36]. To address this issue, we selectively silenced the M1, M2 and M3 genes using siRNA. Our results suggest that all M1, M2 and M3 receptors participate in CSE-induced expression of myofibroblast markers.
In summary, we have demonstrated that cigarette smoke, which is the main risk factor of COPD and participates in lung remodelling, may increase the expression of the myofibroblast markers collagen type I and α-SMA through the activation of a non-neuronal cholinergic system, which is attenuated by the anticholinergic aclidinium bromide. Therefore, results observed in this work support that aclidinium bromide may play a role in regulating the peribronchiolar fibrotic remodelling of COPD in addition to the classical bronchodilator activity.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This work was supported by Almirall S.A. (Barcelona, Spain) and, in part, by grants SAF2011-26443 (to J. Cortijo), SAF2009-08913 (to E.J. Morcillo), FIS (Miguel Servet CP11/00293 (to J. Milara)), CIBERES (CB06/06/0027) and research grants from Regional Government (Prometeo/2008/045, “Generalitat Valenciana”; research grants from “Generalitat Valencia” AP-178/11 (to J. Milara)). T. Peiró received a research grant from Conselleria de Educacion, Generalitat Valenciana ACIF/2010/114. We thank C. Bryant from Complete Medical Communications (Macclesfield, UK) who provided copy-editing support funded by Almirall S.A.
Statement of Interest
Conflict of interest information can be found alongside the online version of this article at www.erj.ersjournals.com
- Received January 28, 2012.
- Accepted August 27, 2012.
- ©ERS 2013
References










