





Abstract
The majority of cases of community-acquired pneumonia are caused by Streptococcus pneumoniae and most studies on pneumococcal host interaction are based on cell culture or animal experiments. Thus, little is known about infections in human lung tissue.
Cyclooxygenase-2 and its metabolites play an important regulatory role in lung inflammation. Therefore, we established a pneumococcal infection model on human lung tissue demonstrating mitogen-activated protein kinase (MAPK)-dependent induction of cyclooxygenase-2 and its related metabolites.
In addition to alveolar macrophages and the vascular endothelium, cyclooxygenase-2 was upregulated in alveolar type II but not type I epithelial cells, which was confirmed in lungs of patients suffering from acute pneumonia. Moreover, we demonstrated the expression profile of all four E prostanoid receptors at the mRNA level and showed functionality of the E prostanoid4 receptor by cyclic adenosine monophosphate production. Additionally, in comparison to previous studies, cyclooxygenase-2/prostaglandin E2 related pro- and anti-inflammatory mediator regulation was partly confirmed in human lung tissue after pneumococcal infection.
Overall, cell type-specific and MAPK-dependent cyclooxygenase-2 expression and prostaglandin E2 formation in human lung tissue may play an important role in the early phase of pneumococcal infections.
Pneumonia is one of the most common causes of death worldwide [1] and Streptococcus pneumoniae is the most frequently isolated pathogen in pneumonia patients [2]. Vaccines are available but only provide protection against selected serotypes [3]. Moreover, the number of resistant strains has increased [4] and multi drug-resistant serotypes have emerged [5]. Thus, it is important to investigate pathogen–host interactions as the basis for the design of novel adjunctive treatment approaches.
Most studies on host–pathogen interaction, including pneumococcal infections, have been based on cultured and, frequently, immortalised cell lines or animal experiments [6]. These models represent an integral part of current pneumonia research, but the obvious limitations of the models should be considered [7, 8], and the principal investigation or, at least, the validation of results should be performed in original human material whenever possible.
Therefore, we established a S. pneumoniae ex vivo infection model of human lung tissue, which was obtained from patients undergoing lung resection due to lung cancer.
Cyclooxygenase (COX)-derived generation of prostaglandins (PGs) is suspected to play an important regulatory role in the innate immunity of the lung [9]. COX-1 is generally believed to be constitutively expressed, whereas COX-2 is the inducible isoform [10]. Induction is caused by a variety of stimuli including bacteria, viruses and cyto-/chemokines [11–14]. They can lead to the activation of mitogen-activated protein kinases (MAPKs), which contribute to various important cell functions, including the regulation of inflammation. For example, p38 MAPK-dependent induction of COX-2 by bacteria and consecutive PG formation has been shown in cultured bronchial epithelium [11–13].
One of the most prominent PGs is PGE2, which has been shown to contribute to the inflammatory response of various cell types [15–18]. PGE2 seems to signal through four distinct G protein-coupled E prostanoid receptors, EP1–EP4 [9].
In this study, we used freshly isolated human lung tissue which was infected ex vivo with S. pneumoniae. We hypothesised that pneumococcal infection may induce COX-2 as well as related formation of arachidonic acid-derived products and investigated the cell type-specific COX-2 expression pattern and the underlying signal transduction pathways. The results clearly establish that in the alveolus, next to alveolar macrophages (AMs), COX-2 is predominantly induced in type II, but not type I, alveolar epithelial cells (AECs). A similar expression pattern of COX-2 in AECs was also demonstrated in lungs of patients who suffered from acute pneumonia. Inhibition of p38 MAPK or extracellular regulated kinase (ERK)1/2 abolished the induction of COX-2 and subsequent release of PGE2. In human lung tissue, activation of EP4 induced a significant increase of cyclic adenosine monophosphate (cAMP). Overall, this study is, to our knowledge, the first to establish the robust effects of pneumococcal infection on prostanoid metabolism in primary human lung tissue.
MATERIALS AND METHODS
For a detailed description of methods, see the online supplementary material.
Materials
PGE2 and PGE1 alcohol (PGE1-OH) were purchased from the Cayman Chemical Company (Ann Arbor, MI, USA). 3-isobutyl-1-methylxanthine and forskolin were purchased from Merck (Darmstadt, Germany). Trichloroacetic acid was obtained from Sigma-Aldrich (Munich, Germany). U0126, SB202190 and NS-398 were purchased from Calbiochem (Merck, Bad Soden, Germany). Tumour necrosis factor (TNF)-α and interleukin (IL)-1β were obtained from R&D Systems (Wiesbaden, Germany). All other chemicals used were of analytical grade and obtained from commercial sources.
Bacterial strains
Encapsulated S. pneumoniae D39 serotype 2 (NCTC7466) (gift from S. Hammerschmidt, University of Greifswald, Greifswald, Germany) was cultured as described previously [19].
Human lung tissue
Fresh lung explants were obtained from patients undergoing lung resection at local thoracic surgery clinics. Additionally, lung tissue samples from three patients with acute pneumonia and one control patient were randomly selected from routine cases. Written informed consent was obtained from all patients and the study was approved by the ethics committee at the Charité Clinic, Berlin, Germany (protocol numbers EA2/050/08 and EA2/023/07). For infection with S. pneumoniae, tumour free normal lung tissue was stamped into small cylinders (∼8×8 mm) and weighed. Specimens were incubated for 24 h in RPMI 1640 with 10% (vol/vol) heat-inactivated fetal calf serum (except for bacterial growth) prior to experimental infection. 200 μL prepared control or infection medium was injected per 100 mg tissue, thereby assuring thorough stimulation of the tissue. The lungs were processed for further analysis at a number of time points.
Colony forming unit assay
Human lung tissue was infected with S. pneumoniae (102 cfu·mL−1) for 0 h (controlled load directly after infection) and 8 h. Afterwards, specimens were disrupted using the FastPrep-24 homogeniser (MP Biomedicals, Heidelberg, Germany) and the supernatants were plated on Columbia agar. Bacterial colonies were counted and cfu·g−1 lung tissue were calculated.
Western blotting
Western blotting was performed as described previously [20]. The antibodies used were against COX-1, COX-2, ERK2, actin (Santa Cruz Biotechnology, Paso Robles, CA, USA), EP4 receptor (Cayman Chemical Co.), phosphorylated ERK or p38 (Cell Signaling, Danvers, MA, USA). ERK2 or actin were used as loading controls. Proteins were visualised by secondary IRDye 800- or Cy5.5-labelled antibodies with an Odyssey infrared scanner (LI-COR Inc., Bad Homburg, Germany).
Immunohistochemistry and confocal immunofluorescence
After infection of human lung tissue, specimens were fixed in formalin, embedded in paraffin and processed for histology. Primary COX-2 antibody (Cayman Chemical Co.) was incubated overnight and detected with the Super Sensitive Link Label Detection System (Biogenex, Fremont, CA, USA). Mayer's haematoxylin (Carl Roth, Karlsruhe, Germany) was used to counterstain nuclei and slides were mounted with Aquatex (Merck). Antibody specificity was verified using blocking peptide. For confocal imaging, COX-2 was labelled with Alexa Fluor 488 or 594 (Invitrogen, Darmstadt, Germany; all diluted 1:2,000). Type I epithelial cells were detected with antibodies against caveolin 1 (Santa Cruz Biotechnology), whereas type II pneumocytes were labelled with antibodies against surfactant protein C precursor (pro-SP-C) (Millipore, Billerica, MA, USA). S. pneumoniae was stained with antibodies against S. pneumoniae (donation by S. Hammerschmidt) and slides were analysed using a Zeiss Axioskop 2 mot [21] or a Zeiss LSM 780 confocal microscope (Zeiss, Jena, Germany).
PGE2 ELISA
Human lung tissue specimens were infected with S. pneumoniae as indicated. PGE2 was measured in the supernatant according to manufacturer's instructions (R&D Systems, Minneapolis, MN, USA).
Mass spectrometry
Human lung tissue specimens were infected with S. pneumoniae for 16 h and supernatants were analysed for free and lipid matrix-bound eicosanoids as described elsewhere [22].
Quantitative PCR
Total RNA extraction was performed using TRIzol (Invitrogen). After tissue disruption, quantitative PCR (20 μL reaction volume or CFX96, 10 μL reaction volume; iCycler, Bio-Rad, Munich, Germany) was carried out. For the calculation of EP receptor copy numbers, plasmids with cloned cDNAs coding for EP-receptors and glyceraldehyde-3-phosphate dehydrogenase were used as templates to prepare standard curves with defined copy numbers.
cAMP ELISA
For cAMP measurement, lung tissue was weighed and processed in trichloroacetic acid. Samples were homogenised and cAMP ELISA was carried out according to manufacturer's instructions (Cayman Chemical Co.).
Bioplex protein array system
For determination of cytokine release, human lung tissue was infected with 106 cfu·mL−1 S. pneumoniae for 16 h. Supernatants were collected and cytokine release was analysed using the Bioplex Protein Array System with beads specific for TNF-α, IL-1β, IL-17, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1 receptor antagonist (IL-1RA), IL-10, IL-15, platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), IL-6, IL-8 and macrophage inflammatory protein (MIP)-1α (BioRad, Hercules, CA, USA).
Statistical methods
Data are presented as mean±sem of at least three separate experiments. The one-tailed Wilcoxon signed rank test was used to test for significant differences between means. A p-value of <0.05 was considered significant.
RESULTS
S. pneumoniae-induced expression of COX-2 in human lung tissue
To assure reproducible pneumococcal infection of human lung tissue, bacterial growth was measured in eight lung tissue samples demonstrating strong and regular bacterial replication within 8 h (fig. 1). No bacterial growth was detected in serum-free culture medium alone (data not shown), confirming that the growth of pneumococci is dependent on factors released by the human lung tissue.

Growth of Streptococcus pneumoniae in human lung tissue. Human lung tissue was infected with S. pneumoniae (strain D39; 102 cfu·mL−1) and colony forming units were determined at 0 h and 8 h. Data are presented as mean±sem of eight different samples. **: p<0.01 versus 0 h of growth.
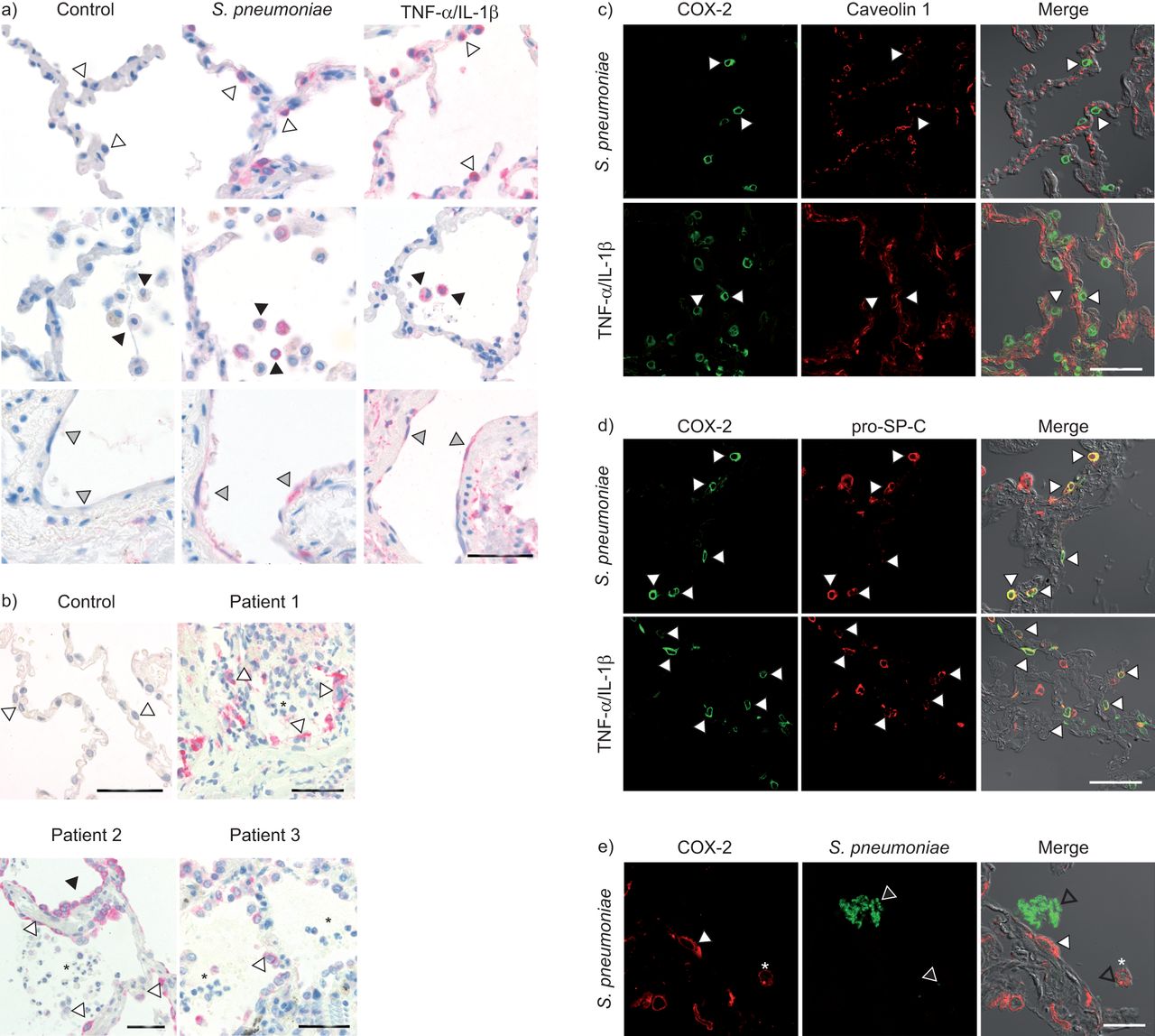
After infection, we observed a strong and time-dependent COX-2 protein induction, whereas constitutively expressed COX-1 remained unaffected in all human lung tissue samples investigated (fig. 2a (four samples) and b (three samples)). In preliminary experiments, we found that compared to the serotype 2 (D39) strain, invasive and noninvasive clinical pneumococcal isolates also induced COX-2 expression in a similar pattern (data not shown). In the next step, immunohistochemistry was performed to study the cell type-specific expression of COX-2, revealing induction mainly in AECs, AMs and in vascular endothelial cells after pneumococcal infection (10 samples) as well as after TNF-α/IL-1β (five samples) treatment in human lung tissue (fig. 3a). Interestingly, we also detected strong COX-2 expression in AECs at inflammatory sites in the lung tissue of three patients suffering from acute pneumonia (fig. 3b). Specificity of COX-2 staining was confirmed by pre-incubation of the corresponding blocking peptide with infected lung sections (data not shown). Double staining of tissue sections with anti-caveolin 1 (type I cells) [23] or anti-pro-SP-C (type II cells) on S. pneumoniae-infected and TNF-α/IL-1β-stimulated tissue was used to confirm the AEC type for COX-2 expression. Notably, despite the stimulus used, almost all COX-2-positive AECs were clearly labelled for pro-SP-C and thus identified as type II cells, whereas no COX-2 staining was observed in type I cells (fig. 3c and d). These observations were confirmed in the lung tissue of three pneumonia patients (data not shown). Interestingly, COX-2 expression was not only detected in cells directly faced to pneumococci, but also in cells without attachment of pneumococci (fig. 3e shown in five samples).

Regulation of cyclooxygenase (COX)-1 and COX-2 by Streptococcus pneumoniae in human lung tissue. Human lung tissue was infected for a) 24 h or b) the indicated time periods with 106 cfu·mL−1 S. pneumoniae. COX-1 and COX-2 protein expression was analysed by Western blot. Representative experiments from a) four and b) three different samples are shown.

a) Human lung tissue was stimulated with Streptococcus pneumoniae or tumour necrosis factor (TNF)-α/interleukin (IL)-1β. Immunohistochemical staining revealed cyclooxygenase (COX)-2 expression (indicated by red colour) in alveolar epithelial cells (AECs) (open arrowheads), alveolar macrophages (black arrowheads) and endothelial cells (grey arrowheads) in S. pneumoniae as well as in TNF-α/IL-1β-stimulated tissue, whereas no staining was observed in lungs stimulated with medium only (control). Scale bars=50 μm. b) In tissues of patients with acute pneumonia, COX-2 was also found to be expressed in AECs (open arrowheads). Acute inflammation can clearly be seen by the infiltration of neutrophils into the alveolar space (black asterisks). Black arrowheads indicate stained distal bronchial epithelium (in patient 2). Healthy, noninflamed control tissue was obtained from patients who underwent tumour resection. No COX-2 expression could be found in control tissue. Prior incubation of the primary antibody with its corresponding blocking peptide abolished staining, indicating antibody specificity (data not shown). Scale bars=50 μm. c) Multicolour immunofluorescent COX-2 (green channel) and caveolin 1 (red channel) staining of S. pneumoniae and TNF-α/IL-1β-stimulated tissue and subsequent confocal imaging revealed no colocalisation of COX-2 and caveolin 1 expression. This implies that COX-2 is not induced in type I AECs. Representative cells are indicated by white arrowheads in each channel. Scale bars=50 μm. d) In contrast, both S. pneumoniae and TNF-α/IL-1β-stimulated lungs revealed COX-2 expression (green channel) in pro-surfactant protein C (pro-SP-C) positive cells (red channel), indicating COX-2 expression in type II AEC. Representative cells with colocalisation of COX-2 and pro-SP-C expression (yellow in merged channels) are indicated by white arrowheads in each channel. Merged panels demonstrate lung structure by using differential interference contrast. Scale bars=50 μm. e) Colonies of S. pneumoniae (green channel, open arrowheads) were detected in the alveolar space adjacent to COX-2- positive (red channel, white arrowhead) type II AECs as well as attached to alveolar macrophages (white asterisk). Scale bars=20 μm. Merged panels demonstrate lung structure by using differential interference contrast microscopy. Representative figures of 10 S. pneumoniae-infected lungs, of five TNF-α/IL-1β-stimulated lungs and of three different patients with acute pneumonia are shown in a)–d). Representative figures of five separate experiments are shown in e).
COX-2 induced PGE2 formation is p38 and ERK dependent
Six human lung tissue samples were infected with S. pneumoniae in the presence or absence of the COX-2 inhibitor NS-398 and PGE2 liberation was analysed. PGE2 levels significantly increased 16–24 h after infection and were abolished by NS-398 (fig. 4a). Previous in vitro studies using cultured lung epithelial cell lines [11–13] indicated that COX-2 expression is dependent on MAPK activity. We observed increased p38 phosphorylation in presence of pneumococci whereas ERK phosphorylation was already detected in uninfected human lung tissue (fig. 4b, five samples). Pre-treatment of seven lung tissue samples with the ERK inhibitor U0126 or p38 inhibitor SB202190 abolished COX-2 protein expression (fig. 4c) as well as PGE2 release in the supernatant (fig. 4d).

Time-dependent cyclooxygenase (COX)-2, extracellular-signal regulated kinase (ERK) 1/2 and p38 mitogen activated protein kinase (MAPK)-mediated induction of prostaglandin (PG)E2 by Streptococcus pneumoniae. a) and b) human lung tissue was infected with 106 cfu·mL−1 S. pneumoniae or pre-treated with selective COX-2 inhibitor (NS-398, 10 μM, 1 h) and infected for the indicated time periods. a) Time-dependent PGE2 release was inhibited by COX-2 inhibition as measured by ELISA. b) Western blots demonstrate activation of p38 MAPK by S. pneumoniae, whereas ERK phosphorylation was already visible in uninfected tissue. c) and d) tissue was pre-treated for 1 h with ERK MAPK inhibitor (U0126 10 μM) or p38 MAPK inhibitor (SB202190 10 μM) and infected with 106 cfu·mL−1 S. pneumoniae for 8 h. c) COX-2 and d) PGE2 induction was suppressed by MAPK inhibitors as shown by Western blot and ELISA, respectively. Data are presented as mean±sem of a) six and d) seven different samples. Representatives of b) five and c) seven blots are shown. P-p38: phosphorylated p38; P-ERK: phosphorylated ERK. *: p<0.05 versus control; **: p<0.01 versus control; #: p<0.05 versus infected tissue without pre-incubation with inhibitor; ##: p<0.01 versus infected tissue without pre-incubation with inhibitor.
COX-2 expression is not regulated by a positive feedback loop
In mouse lung fibroblasts, using PGE2 stimulation, a positive feedback loop was demonstrated by Vichai et al. [24], indicating that COX-2-related PGE2 production further fostered COX-2 expression. Therefore, we measured COX-2 expression after pre-incubation of five lung tissue samples with COX-2 inhibitor NS-398 for 1 h (fig. 5). However, in human lung tissue, S. pneumoniae-induced COX-2 expression was unaffected by prior inhibition of COX-2 (fig. 5a). Since exposure of three lung tissue samples to different doses of PGE2 failed to affect COX-2 protein expression, no positive feedback loop, as presented in mouse fibroblasts, was found in intact human lung tissue. TNF-α/IL-1β stimulation served as positive control (fig. 5b).

Western blot of Streptococcus pneumoniae-induced cyclooxygenase (COX)-2 shows no feedback regulation. a) Human lung tissue was infected with 106 cfu·mL−1 S. pneumoniae or pre-treated with a selective COX-2 inhibitor (NS-398 10 μM, 1 h) prior to infection. COX-2 induction was analysed by Western blotting showing no inhibition of COX-2 expression. b) Lung tissue was incubated for 24 h with the indicated concentrations of prostaglandin (PG)E2 or with tumour necrosis factor (TNF)-α (100 ng·mL−1) and interleukin (IL)-1β (10 ng·mL−1). Representatives of a) five and b) three blots are shown.
Induction of COX-2 metabolites by S. pneumoniae
In principle, a variety of highly biologically active mediators can be produced via COX-2 [25]. Therefore, five human lung tissue samples were infected with or without pre-treatment of the COX-2 inhibitor NS-398 and mass spectrometry was performed to detect 6-keto-PGF1α, thromboxane (TX)B2, PGE2, PGH2, PGE2 ethanolamide, PGF2α and 8-iso-PGE1. An increase after infection and a decrease after NS-398 pre-incubation was measured for PGE2, 6-keto-PGF1α and TXB2 (fig. 6a–c), whereas other metabolites remained unchanged (data not shown).

Induction of cyclooxygenase (COX)-2 metabolites by Streptococcus pneumoniae. Human lung tissue was pre-incubated with selective COX-2 inhibitor (NS-398; 10 μM) for 1 h before infection with 106 cfu·mL−1 S. pneumoniae for 16 h. a) Prostaglandin (PG)E2, b) 6-keto-PGF1α and c) thromboxane (TX)B2 release in the supernatant was measured using mass spectrometry. Data are presented as mean±sem of five different samples. ND: not detected. *: p<0.05 versus control; #: p<0.05 versus infected tissue without pre-incubation with NS-398.
EP receptor expression profile in human lung tissue
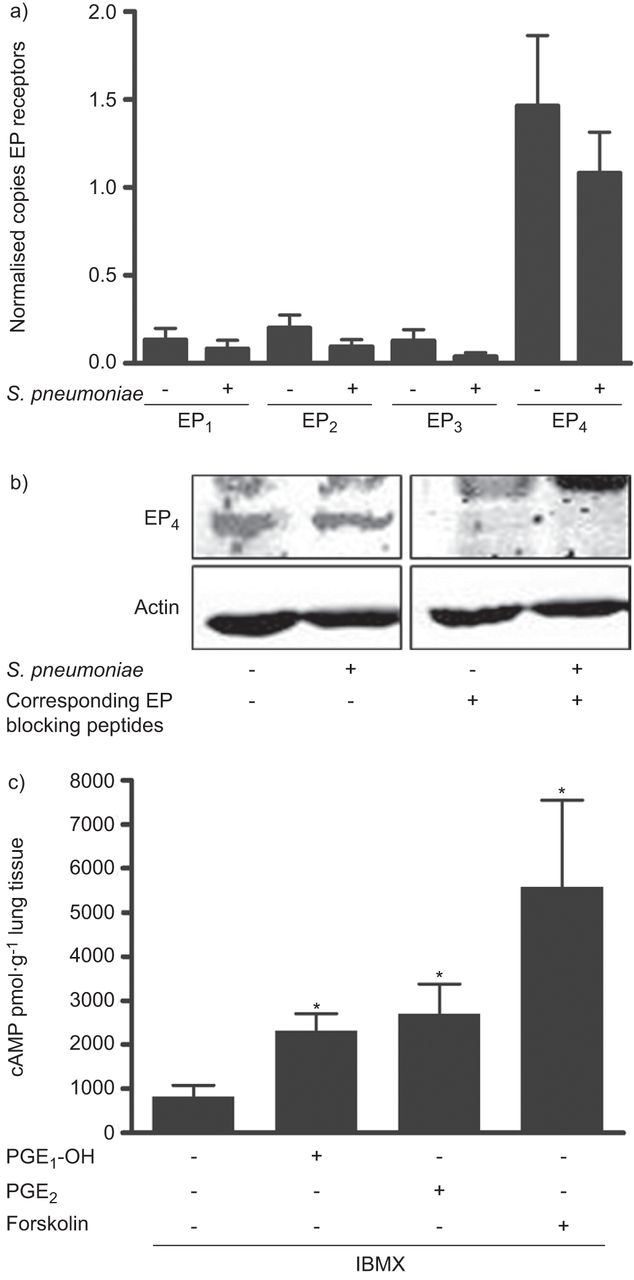
PGE2 signalling is mainly transduced by the prostanoid receptors EP1–4 [9]. Therefore, we investigated their regulation in three lung tissue samples after infection with S. pneumoniae. Low copy counts of mRNA were detected for EP1–3 whereas higher levels were observed for EP4 (fig. 7a). EP4 expression was also demonstrated at the protein level in three lung tissue samples (fig. 7b). However, within the investigated timeframe of 18–24 h pneumococcal infection neither changed receptor expression levels on the mRNA nor on the protein level (fig. 7a and b). Since EP4 activation leads to cAMP induction we analysed receptor functionality by the EP4 agonist PGE1-OH. Selective EP4 activation as well as addition of PGE2 or forskolin significantly increased intracellular cAMP levels in six human lung tissue samples (fig. 7c).

E prostanoid (EP) receptor expression pattern in human lung tissue. Human lung tissue was infected for a) 18 h or b) 24 h with 106 cfu·mL−1 Streptococcus pneumoniae. a) EP1–4 mRNA was analysed using quantitative PCR, demonstrating highest expression levels for EP4. b) EP4 protein was detected by Western blotting showing no induction after bacterial infection. Antibody specificity was verified using corresponding blocking peptides. c) Human lung tissue was pre-treated with the nonspecific phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) (20 μM) for 2 h and stimulated with a specific EP4 receptor agonist (prostaglandin E1 alcohol (PGE1-OH)) (10 μM), prostaglandin (PG)E2 (PGE2) (10 μM) or forskolin (20 μM) for 30 min. Cyclic adenosine monophosphate formation was determined by ELISA demonstrating a clear increase in human lung tissue. Data are presented as mean±sem of a) three and c) six different samples. Representative samples of three blots are shown in b). *: p<0.05 versus control.
COX-2 regulation of S. pneumoniae-induced cytokine release in human lung tissue
PGE2 induction has previously been demonstrated to significantly contribute to chemo-/cytokine regulation [15–18]. Therefore, we analysed five infected human lung tissue samples for chemo-/cytokine release in the presence or absence of COX-2 inhibitor NS-398. Pneumococci significantly induced TNF-α, IL-1β, PDGF, GM-CSF, IL-17, IL-10, and IL-15 liberation (fig. 8a–g). Furthermore, IL-1RA was significantly increased, whereas VEGF, IL-6, IL-8 and MIP-1α levels remained unchanged following pneumococcal infection (data not shown). As expected, COX-2 inhibition reduced PGE2 in four lung tissue samples and resulted in a significant increase of PDGF, and slight increases of TNF-α (p<0.09) and GM-CSF (p<0.15) expression, but displayed no effect on other chemo-/cytokines in the same experiments (fig. 8a–h).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Streptococcus pneumoniae induces pro- and anti-inflammatory cytokines in human lung tissue. Human lung tissue was pre-treated with and without selective cyclooxygenase (COX)-2 inhibitor (NS-398; 10 μM) for 1 h and then infected with 106 cfu·mL−1 S. pneumoniae for 16 h. a) Tumour necrosis factor (TNF)-α, b) interleukin (IL)-1β, c) granulocyte macrophage-colony stimulating factor (GM-CSF), d) platelet-derived growth factor (PDGF), e) IL-10, f) IL-15 and g) IL-17 release in the supernatant was measured using Bioplex cytokine assay (BioRad, Hercules, CA, USA), demonstrating a slight increase for TNF-α and a significant increase for platelet-derived growth factor after COX-2 inhibition. h) Prostaglandin (PG)E2 release in the same samples was used as positive control. Data are presented as mean±sem of five (a–g) and four (h) different samples. *: p<0.05 versus control; #: p<0.05 versus infected tissue without pre-incubation with NS-398.
DISCUSSION
COX-2-derived metabolites are important regulators of inflammation [25]. Studies using cultured lung cell lines [11–13] and mice [26, 27] indicated a prominent immunomodulatory role of COX-2 in pneumonia. Since there is virtually no study that has systematically analysed the biology of COX-2 in the inflamed human lung, we used an ex vivo model of pneumococcal infection [28] of freshly isolated peripheral human lung tissue to explore cell type-specific COX-2 regulation and function. An important advantage of our model system is that the different lung cell types are still organised into the unique lung architecture and cell-specific behaviour can therefore be studied. Resident cells including AMs are still present in the tissue and are capable of contributing to the observed response as shown previously in a similar model [28]. However, this model does not allow for investigations of aspects of immunity such as the recruitment of immune cells from the blood. Moreover, the disconnection of lung tissue from the bloodstream, oxygen supply and gas exchange can only partly be compensated for by adaption of cell culture conditions. As a consequence, the inflammatory response might be influenced by, for example, cell death by apoptosis and necrosis. Therefore, a timeframe of up to 24 h has been used where neither lactate dehydrogenase release nor increase of caspase-3 activation was observed (data not shown). Furthermore, the infection route of our infection model varies from the natural one since bacteria are directly injected into the lung tissue, allowing bacteria to bypass the route from the trachea to the alveolus. Although this method does not reflect the natural infection route it nevertheless reproducibly allows the study of the direct interaction of bacteria with the alveolar compartment, revealing the inflammatory response of the resident cell population. However, the validity of the results obtained from this model should be compared with samples from pneumonia patients whenever possible, which could also be performed in the present study. Additionally, the availability of such human lung tissue samples is limited, leading to small experimental sample sizes. This is why robust biological signals are required for statistically significant results. We found significant and tissue-dependent growth of S. pneumoniae in human lungs that induced the expression of the COX-2/PGE2/cAMP axis, thereby partly regulating bacteria-induced chemo-/cytokine expression. The panel of mediators induced is comparable to those in studies having investigated in vitro infected bronchial epithelial cell lines [29].
The results of this study established that pneumococci are capable of inducing strong expression of COX-2, whereas COX-1 was constitutively expressed and remained unaffected in the infected lung. Immunohistochemistry revealed COX-2 expression in AMs, AECs and the vascular endothelium. The relevance of these findings was further supported by the positive staining of AECs in lungs of patients with acute pneumonia. Interestingly, within the alveolar epithelium, induction of COX-2 was almost exclusively seen in type II but not in type I AECs. Since TNF-α/IL-1β exposure of lung tissue produced the same pattern, it appears reasonable to assume that type II cells are, in general, important alveolar epithelial pacemakers of inflammation. The pro-inflammatory environment after pneumococcal infection or cytokine treatment also fostered COX-2 expression in alveolar macrophages and lung endothelium. Apart from these cell types we occasionally observed COX-2-positive stromal cells, which were neither caveolin 1 nor pro-SP-C positive. Notably, a comparable expression pattern of COX-2 was seen in cynomolgus monkeys with acute severe pneumonia in vivo [30]. We detected S. pneumoniae in areas where no COX-2 positive cells were found and vice versa. This finding suggests that COX-2 induction is not necessarily related to cellular attachment of S. pneumoniae. Factors released in the supernatant either from pneumococci or from the lung tissue might foster COX-2 expression which is supported by our data from TNF-α/IL-1β-stimulated lungs also showing strong COX-2 expression in the same cell types.
In infected lung tissue the chemical inhibitor NS-398 almost completely blocked the release of PGE2, a major COX-2 product. In addition, we observed production of 6-keto-PGF1α and TXB2. However, no release of other prostanoids was found in our experiments. As evidenced by previous studies, PGE2 seems to be the major prostanoid produced by human lung cells [31]. Since PGE2 produced by lung cells downregulates neutrophil responses it may dampen the pro-inflammatory potential of neutrophils, thereby preventing epithelial damage [31].
In vitro experiments have previously established that induction of COX-2 upon bacterial infection is dependent on MAPK [11–13]. In line with the study of Xu et al. [28], we observed activation of p38 MAPK during pneumococcal infection in human lungs, whereas ERK was already activated in control lungs. Inhibition of both kinases suppressed S. pneumoniae-induced COX-2 induction and subsequent PGE2 release in our human lung infection model. In some studies PGE2 was shown to regulate COX-2 expression in a positive feedback loop, whereas other studies even reported a repressive effect of PGE2 [24]. In human lungs, neither inhibition of COX-2 nor the direct stimulation with different doses of PGE2 had any effect on COX-2, demonstrating that the feedback loop is most probably not present in human lung tissue.
PGE2 exerts its effects through four different E prostanoid receptors (EP1–EP4) which show a cell type-specific expression [9] and, previously, we have demonstrated that Moraxella catarrhalis infection led to an upregulation of EP2 and EP4 protein in bronchial epithelium [13]. Additionally, in S. pneumoniae-infected mice, deficiency of EP3 was beneficial for bacterial clearance and survival, underlining the role of EPs in bacterial infection [32]. It is well established that activation of EP receptors by PGE2 subsequently changes the level of intracellular cAMP, which, finally, might contribute to the inflammatory reaction [9]. However, neither EP receptor expression nor cAMP generation has been shown in pneumococcal-infected human lung tissue so far. Therefore, we investigated EP mRNA expression, demonstrating low EP1–EP3 levels and high levels for EP4. Although pneumococcal infection of peripheral lung tissue did not alter constitutive EP4 receptor expression on an mRNA nor a protein level, specific receptor agonists for EP4 induced a significant increase of cAMP in the lung tissue, which might influence the regulation of inflammatory mediators after S. pneumoniae infection.
Several studies have demonstrated strong chemo-/cytokine induction after infection with pro-inflammatory stimuli, or even with S. pneumoniae, in vitro as well as in vivo [15–18, 29, 33]. Similarly, pneumococcal infection of human lung tissue also revealed significant induction of TNF-α, IL-1β, GM-CSF, PDGF, IL-10, IL-1RA, IL-15 and IL-17. However, other expected mediators, such as IL-6, IL-8, MIP-1α and VEGF, failed to be induced, indicating either cell-specific expression in the bronchial compartment that is not present in this model or differences in the mediator regulation in human lung tissue per se. Furthermore, it has been shown that COX-2/PGE2 may contribute to the inflammatory control by inducing IL-10, or inhibiting GM-CSF, TNF-α, IL-12 or PDGF expression in vitro [15–18, 34]. In line with these studies COX-2 inhibition led to a significant induction of PDGF [18], but only slight increases for TNF-α [16, 34] and GM-CSF [15]. Since the effects of PGE2 have been shown in isolated cell types of different origin, small or counteracting regulation of cytokines in the alveolar compartment might not be measureable in sum signals from whole lung tissue. Moreover, another possibility of the slight effects measured for COX-2 inhibition might be that PGE2 is primarily directed against invading immune cells, which are not represented in this organ model.
In summary, using peripheral human lung tissue infected ex vivo with S. pneumoniae we demonstrated MAPK-related induction of COX-2 and subsequent PGE2 release. In the alveolus COX-2 is induced in type II, but not type I alveolar epithelial cells as well as in AMs and vascular endothelium. In human lung tissue, activation of EP4 increased cAMP. PGE2 production by lung cells may regulate immune cell function and contribute to the control of inflammatory mediator production in pneumococcal pneumonia of the human lung. Further studies using human lung tissue are needed to understand the complexity of lung inflammation, in particular, the role of type II pneumocytes, and to verify results obtained with cell lines and animal models.
Acknowledgments
The excellent technical assistance of D. Stoll and A. Kühn, as well as the proof reading of J. Lienau (all at Department of Internal Medicine/Infectious Diseases and Pulmonary Medicine, Berlin, Germany), is greatly appreciated. Part of this work will be included in the doctoral thesis of K.V. Szymanski.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This study was supported by the Transregional Collaborative Research Center SFB-TR84 of the Deutsche Forschungsgemeinschaft (grants C5 to A.C. Hocke and S. Hippenstiel, Z1a to A.C. Hocke and Z1b to A.D. Gruber) and the German Federal Ministry of Education and Research (grant C8 to A.C. Hocke; PROGRESS – Pneumonia Research Network on Genetic Resistance and Susceptibility for the Evolution of Severe Sepsis).
Statement of Interest
None declared.
- Received October 26, 2011.
- Accepted February 23, 2012.
- ©ERS 2012
REFERENCES










