





Abstract
Pulmonary hypertension (PH) is a severe and progressive disease characterised by high pulmonary artery pressure, usually culminating in right heart failure. Current therapeutic approaches in PH largely provide symptomatic relief while the prognosis rate is lower due to the lack of specific molecular targets and the involvement of several factors in the development of PH. Numerous studies have suggested a crucial role of matrix metalloproteinase (MMP) axis during development and disease states, specifically with regard to extracellular matrix remodelling and vascular homeostasis. Increased MMP activity has been demonstrated in experimental animal models of PH, and MMP inhibition has been shown to either attenuate or enhance vascular remodelling. Moreover, several studies emphasise that restoration of deregulated MMPs to physiological MMP/tissue inhibitor of MMPs ratios would potentiate reverse remodelling in PH. This article will highlight the pathophysiological role of MMPs in vascular remodelling and the establishment of PH. In particular, we will focus on the MMP expression and regulation in pulmonary vasculature and pulmonary vascular remodelling. We will also provide an overview of recent clinical and experimental findings and their impact on achieving maximum reversal of PH, as well as current issues and future perspectives.
PULMONARY HYPERTENSION
Pulmonary hypertension (PH) is not a disease per se but rather a pathophysiological parameter defined by a mean pulmonary arterial pressure (Ppa) exceeding the upper limits of normal, i.e. 25 mmHg at rest [1]. PH occurs in a variety of clinical situations and is associated with a broad spectrum of histological patterns and abnormalities. A new classification of clinical PH, which was revised recently, designated five categories that are distinctive in their clinical presentation, diagnostic findings and response to treatment. Among these, group 1 comprises a group of diverse diseases termed pulmonary arterial hypertension (PAH). The other four main clinical groups of PH include pulmonary veno-occlusive disease (group 1’), PH due to left heart disease (group 2), PH due to lung diseases (group 3), chronic thromboembolic pulmonary hypertension (group 4) and PH with unclear or multifactorial aetiologies (group 5) [2].SERIES “MATRIX METALLOPROTEINASES IN LUNG HEALTH AND DISEASE”Edited by J. Müller-Quernheim and O. EickelbergNumber 9 in this Series
PAH is characterised by endothelial injury, persistent vasoconstriction and obliterative remodelling of pulmonary arteries, which subsequently reduces the cross-sectional area of pulmonary microvasculature and increases the vascular resistance. Subsequent elevation of Ppa further increases the right ventricular afterload leading to right ventricular hypertrophy and heart failure [3]. Decades of research indicate the central role of pulmonary endothelial cell dysfunction in the initiation and progression of pulmonary vascular remodelling, commencing with the imbalance in production of vasoactive mediators (such as nitric oxide (NO), prostacyclin, endothelin and others). The progressive phase of PAH involves formation of intimal and plexiform lesions, endothelial apoptosis, medial thickening, adventitial thickening and increased extracellular matrix (ECM) turnover, as well as accumulation of ECM proteins. These events eventually lead to capillary rarification due to pulmonary arteriolar occlusion and microvascular degeneration of distal pulmonary vasculature [3–5]. Of the aforementioned multiple vascular abnormalities involved in the establishment of PAH, imbalance in ECM synthesis and degradation contributes to the complexity of the pathological remodelling process and worsens treatment outcome [6, 7].
The structural alterations in pulmonary vasculature are arbitrated by activated vascular cells, which acquire and exhibit hyperproliferative, migratory and invasive capabilities. These phenotypic abnormalities may be facilitated by ECM degrading enzymes in response to different pathological stimuli [3].
ECM OF VASCULATURE
The vascular ECM is a complex meshwork of matrix molecules, which are assembled in an orchestrated manner that contributes to the structural and functional integrity of the vasculature. The ECM provides mechanical strength, elasticity and compressibility to the vessels. In addition, the ECM also provides a complex microenvironment facilitating cell–cell and cell–ECM crosstalk to regulate cell migration, proliferation and differentiation to eventually sustain vascular homeostasis [8]. The vessel wall that encloses the arterial lumen has a three-layer architecture comprising of the tunica intima, tunica media and tunica adventitia (fig. 1) [9]. Moreover, the pulmonary vascular bed encompasses different cell types, which provide structural and functional integrity to sustain vascular homeostasis.

Architecture of the arterial wall and its extracellular matrix (ECM) components. The vessel wall has a three-layered architecture: the tunica intima (TI), tunica media (TM) and tunica adventitia (TA). The major elements of the arterial wall comprise the cellular component of the vasculature (such as endothelial cells, smooth muscle cells and adventitial fibroblasts (AFs)) and the ECM component of the vessel, which contributes to the framework of a functional vasculature. EC: endothelial cell; BM: basement membrane; IEL: internal elastic lamina; SMC: smooth muscle cells; EEL: external elastic lamina; HSPG-2: heparan sulfate proteoglycan 2; vWF: von Willebrand factor.
The innermost layer, the tunica intima, is composed of a continuous monolayer of flattened vascular endothelial cells (ECs) that line the luminal surface of the arteries and form a tightly regulated semipermeable barrier [10]. Vascular ECs are interconnected by intercellular junction complexes (such as tight junctions, adherens junctions and gap junctions) [11] and rest on the basal lamina of the basement membrane (BM). The supramolecular assembly of the BM is formed by specific interactions between ECM components, such as laminins, type IV collagens, entactins (nidogens), proteoglycans, glycoproteins and different integrin and non-integrin receptors (fig. 1) [12]. Importantly, ECs are known to synthesise a variety of BM and internal elastic lamina (IEL) components and several vasoactive mediators, such as NO, prostacyclin, endothelin (ET)-1, serotonin and thromboxane, which are important for regulating vascular tone [8, 13]. In muscular arteries, IEL can be identified as a fenestrated layer of elastic tissue (elastin) separating the intima and media beneath the sub-endothelial layer which also allows diffusion [8]. The medial layer of the pulmonary artery is profuse and consists of multiple concentric layers of smooth muscle cells (SMCs) and collagen fibres interposed between layers of fenestrated elastic lamina.
In general, arterial SMCs are quiescent, exhibit a differentiated contractile phenotype under normal physiological conditions and constitute the major cell type of the medial layer. Most importantly, SMCs maintain developmental plasticity and are capable of transient phenotypic switching between contractile, synthetic or intermediate phenotypic states [14]. While the contractile phenotype of the SMCs exhibits a low rate of proliferation and synthesis, it renders contractility to the vasculature and regulates the vascular tone in response to vasoactive mediators. In contrast, the synthetic phenotype is characterised by increased proliferation, migration and ECM turnover. It synthesises and secretes different ECM components of the tunica media such as elastin fibres, fibrillar collagens (type I, III and V), elastin, fibronectin, laminin and proteoglycans [15]. These different ECM components have been shown to play a key role in sustaining the SMC phenotype [16]. For example, elastin, one of the major constituents in the ECM of the medial layer [17], is a potent autocrine regulator of vascular SMC activity. Besides rendering resilience and elasticity to the arteries, elastin is critical for stabilisation of the arterial structure by inducing a quiescent contractile state. Several in vitro studies indicate an inverse correlation between elastin expression and SMC proliferation [18–20]. External elastic lamina is a thick fibrous layer of elastic fibres that delimit media from adventitia and is found to be less prominent than IEL. Tunica adventitia is relatively thick in muscular arteries in comparison to elastic arteries and the thickness varies in different parts of the vascular circuit. Adventitial fibroblasts are the predominant cell type of the tunica adventitia and regarded as a critical regulator of vascular wall function in health and disease. Fibroblasts also regulate synthesis and secretion of adventitial ECM components such as collagen types I and III, which constitute the chief ECM content of the adventitia [21]. Besides rendering structural integrity to the vasculature, adventitia has resident immunomodulatory and progenitor cell populations that contribute to the growth and repair processes of the vessel wall [22, 23].
MATRIX METALLOPROTEASES, ADAMALYSINS, SERINE ELASTASES AND THEIR INHIBITORS
Turnover of the ECM is controlled by the balance between proteolytic enzymes, such as matrix metalloproteinases (MMPs), serine elastases and their endogenous inhibitors. MMPs are a family of structurally related, zinc-dependent multifunctional proteases that are either soluble or membrane anchored. MMPs belong to a larger family of proteases known as the metzincin superfamily [24].
MMPs are classified in a number of ways. Historically MMPs are categorised according to their substrate specificity. Based on their substrate specificity, MMPs or matrixins can be subdivided into six groups: 1) interstitial collagenases (MMP-1, MMP-8, MMP-13 and MMP-18); 2) type IV collagenases or gelatinases (MMP-2 and MMP-9); 3) stromelysins (MMP-3, MMP-10, MMP-11 and MMP-19); 4) matrilysins (MMP-7 and MMP-26); 5) transmembrane MMPs (membrane type (MT)-MMPs: MMP-14, MMP-15, MMP-16, MMP-17, MMP-24 and MMP-25); and 6) other MMPs (MMP-12, MMP-20, MMP-21, MMP-22, MMP-23, MMP-27 and MMP-28) [24, 25]. Since several new extracellular and non-extracellular matrix substrates of MMPs are being discovered, MMPs are now commonly referred to by their numerical designations (MMP-1 to MMP-28). Table S1 provides the generic and commonly used group names, taxonomy number, substrate portfolios (both matrix and non-matrix) and cellular sources for MMP types that have been identified to date. However, substrate portfolios for specific MMPs may be incomplete, due to active research occurring in this area.
MMPs typically consist of four well-conserved modular structures: a pro-peptide domain; a catalytic metalloproteinase domain; a linker peptide of variable lengths (also called the “hinge region”); and a hemopexin domain. Exceptions to this are MMP-7 (matrilysin-1), MMP-26 (matrilysin-2) and MMP-23; all of which are missing the linker peptide and the hemopexin domain. In addition, MMP-23 has a unique cysteine-rich domain and an immunoglobulin-like domain after the metalloproteinase domain. Whereas gelatinases like MMP-2 and MMP-9 have three repeats of a fibronectin type II motif in their metalloproteinase domain. This fibronectin type II motif forms a collagen-binding domain allowing for the binding and degradation of type IV collagen or denatured collagen (gelatin) [24, 26].
MMPs are synthesised in an inactive zymogen form with an auto-inhibitory pro-peptide domain (commonly referred to as proMMPs). Activation of these latent MMPs occurs in a coordinated sequence of proteolytic events. The MMP catalytic domain is concealed by the pro-peptide through cysteine–Zn2+ interaction. During MMP activation, proteolytic cleavage of the NH2-terminal sequence of the pro-peptide domain causes a conformation change in the molecule, consequently exposing the Zn2+ binding site of the catalytic domain [27]. Following the initial cleavage of the pro-peptide domain, further autolytic or exogenous cleavages often occur resulting in lower molecular weight active forms of MMP. In active MMP, the catalytic domain containing the Zn2+ binding region is responsible for proteolytic activity. The hemopexin domain confers substrate specificity [26, 27]. Thus, MMPs degrading diverse components of the ECM in vasculature can mediate a wide range of fundamental biological processes including normal embryonic development processes, angiogenesis, reproduction, bone remodelling and tissue repair processes, and are also implicated in several pathological settings [24, 28].
Under physiological conditions, the activities of MMPs are regulated at the level of transcription, translational and post-translational. Another critical control point of MMP activity is through the inhibition of activated enzymes by a group of endogenous inhibitors called tissue inhibitors of MMPs (TIMPs). TIMPs tightly control the expression and activity of MMPs and provide a balancing mechanism to prevent excessive degradation of ECM [27].
The TIMP family is composed of four members, TIMP-1 through TIMP-4. TIMPs have an N- and C-terminal domain of ≈125 and 65 amino acids, respectively, with each containing three conserved disulfide bonds. The N-terminal domain folds as a separate unit and is capable of inhibiting MMPs [29]. TIMPs can form complexes with MMPs in a 1:1 stoichiometric ratio through co-ordination of the Zn2+ of the MMP active site with the amino and carbonyl groups of the TIMP N-terminal cysteine residue. Although TIMPs do not show a high specificity for any particular MMP, there is preferential binding of TIMP-2 with MMP-2 and TIMP-1 with MMP-9. In addition, TIMP-2, -3 and -4, but not TIMP-1, are effective inhibitors of the MT-MMPs. TIMP-3 is unique among the four TIMPs due to its direct binding to ECM proteins, and thus can provide a means for stabilising MMP-TIMP complexes within the interstitial space [24, 30]. TIMP-3 has also been demonstrated to inhibit ADAM (a disintegrin and metalloproteinase), notably ADAM-17 [31]. However, TIMPs are not the only endogenous MMP inhibitors. Indeed, α2-macroglobulin, an abundant plasma protein, is shown to be the major endogenous inhibitor of MMP activity in the plasma. Although TIMPs inhibit MMPs in a reversible manner, α2-macroglobulin/MMP complexes are removed by scavenger receptor-mediated endocytosis and thus play an important role in the irreversible clearance of MMPs [30].
Another member of the metzincin superfamily that is closely related to MMPs is ADAM, also known as the adamalysins or metalloproteinase-like, disintegrin-like, cysteine rich. Adamalysins/ADAMs belongs to the superfamily of zinc-dependent metalloproteinases and consists of two groups: the membrane-anchored ADAMs and the secreted ADAMTSs. At least 40 ADAMs has been described, 25 of which are expressed in humans. Among those, 19 display proteolytic activity [32]. The domain structure of the ADAMs consists of a pro-peptide domain, a metalloprotease domain, a disintegrin domain, a cysteine- rich domain, an endothelial growth factor (EGF)-like domain, a transmembrane domain, and a cytoplasmic tail. Like most proteases, the ADAMs are initially synthesised as enzymatically inactive precursor proteins. This inactive state in most of the ADAMs is due to the interaction of a cysteine residue in the pro-peptide domain with Zn2+ at the catalytic site. For protease activation, this pro-peptide domain is removed by a furin-like convertase or by autocatalysis, depending on the specific ADAM [32, 33]. Next to the prodomain is the metalloproteinase domain. The metalloprotease domain hydrolyses protein substrates such as cytokines and growth factors, as well as their respective receptors. This process is termed ectodomain shedding and ADAM is often termed “sheddases”. The disintegrin domain binds integrin receptors such as α9β1 and, therefore, mediates cell–cell and cell–matrix interactions. The exact functions of the cysteine-rich domain and the EGF-like domain are unclear. However, the cytoplasmic tail containing phosphorylation sites and SH3 binding domains is thought to be involved in signalling and may also serve to assemble a group of cytoplasmic adaptor molecules [33]. However, ADAMTS are characterised by the presence of additional thrombospondin type I motifs in their C-terminal, while EGF-like, transmembrane and cytoplasmic domains are lacking [34].
These different domains composing of ADAM and ADAMTS endow these proteins in multifunctional activities, such as proliferation, migration and angiogenesis. For example, EGF receptor ligands (amphiregulin and heparin-binding EGF) released/shed by ADAM-17 enhance cell proliferation of cancer cells and induce angiogenesis [35]. In accordance, altered expression of specific ADAMs has been implicated in different diseases; their best documented role is in cancer formation and progression [36]. However, to date, no data are available about the roles of those proteins in the pathogenesis of PH.
Among other proteases that degrade ECM, serine elastases are the ones that have been extensively characterised. Serine proteases are the enzymes that cleave peptide bonds in proteins, in which serine serves as the nucleophilic amino acid at the active site. Humans have six elastase genes that encode the structurally similar proteins elastase 1, 2, 2A, 2B, 3A, and 3B [37]. Among several serine elastases, neutrophil elastases secreted by neutrophils and a 23-kDa serine elastase called endogenous vascular elastase (EVE) have been strongly implicated in vascular diseases. EVE is produced by vascular SMCs and degrades elastin and several other ECM proteins [38, 39]. Regulatory control of serine elastase activity is accomplished by the concomitant production of endogenous inhibitors like α1-antitrypsin, α2-macroglobulin and elafin, with the most relevant inhibitor in vasculature being elafin [40].
We have reviewed the physiological and pathophysiological role of MMPs and serine elastases in vasculature with specific focus on the impact of MMPs and serine elastases in the pathogenesis of PH. Furthermore, the pharmacological interventions using MMP inhibitors and outcome in different pre-clinical rodent models and future directions will be discussed.
EXPRESSION AND REGULATION OF MMPs IN VASCULATURE
Vascular cells
Different cell types that constitute the cellular component of the vasculature, including endothelium, SMCs, fibroblasts and infiltrating innate immune cells which infiltrate the vessel wall, are described as the major cellular sources of MMPs in adult vasculature [41–44] (table S1). The diverse functions of MMPs in vasculature mainly depend on its expression and activity. Under physiological and pathological states, biological, mechanical, haemodynamic or neurohormonal stimuli are described to modulate the vascular cell-specific MMP expression and activity [45].
Several studies have demonstrated vascular endothelium as a main source of MMPs during angiogenesis or wound healing processes. ECs were shown to constitutively express detectable levels of MMP-1, MMP-2, TIMP-1 and TIMP-2, but not MMP-9 under basal conditions [41]. Although the basal expression of MMPs was low/undetectable in ECs, several pro-angiogenic factors, pro-inflammatory mediators and organic compounds (e.g. phorbol esters) can induce MMP expression and activate latent MMPs [46]. For example, a vascular endothelial growth factor (VEGF)-dependent increase of MMP-2 activity and substantial reduction of TIMP-1 and TIMP-2 levels was observed in microvascular ECs. VEGF-induced downregulation of TIMP levels subsequently permits activation of pre-existing collagenases, which together could contribute to the endothelial invasion of the BM and interstitial matrix [47].
SMC-specific MMP expression and activity is distinctly modulated by a variety of growth factors and cytokines under pathophysiological conditions. Exposure of cultured SMCs to pro-inflammatory molecules like interleukin (IL)-1α, tumour necrosis factor (TNF)-α, and oxidised low-density protein are shown to significantly increase the expression and activity of MT1-MMPs, which further leads to the increased activation of proMMP-2 [43]. However, another study demonstrated that autocrine expression and activation of transforming growth factor (TGF)-β1 antagonises the platelet-derived growth factor (PDGF)-BB (homodimer of PDGF subunit B/beta polypeptide)-induced upregulation of MMP-2 in SMCs during phenotypic modulation [48], suggesting TGF-β1 promotes contractile phenotype by modulating MMPs. Cumulatively, these findings illustrate the differential regulation of MMP expression by different growth factors and cytokines.
Adventitial fibroblasts are the predominant cell type of the tunica adventitia and are found to be active during growth but are typically quiescent in normal vasculature. In particularly, a significant increase in active MMP-2, TIMP-1 and TIMP-2 production was also observed under hypoxic stimulus when compared to normoxic fibroblasts [44]. It has been shown that the reactive oxygen species (ROS)-induced oxidative stress increases MMP activity and potentially modulates fibroblast proliferation and collagen synthesis [49].
Resident and infiltrating immune cells
Apart from cells of the vasculature, infiltrating inflammatory cells such as cells of the monocyte/macrophage lineage, dendritic cells, neutrophils, mast cells, T-lymphocytes and B-lymphocytes also constitute a major cellular source of the pro-angiogenic proteases, cytokines and growth factors [50].
Monocytes/macrophages
Different in vitro studies have reported synthesis and secretion of MMP-1, MMP-2, MMP-3, MMP-7, MMP-9 and a macrophage-specific metalloelastase (MMP-12) by cultured macrophages ex vivo under either basal or stimulated conditions [51]. Moreover, the exposure of human alveolar macrophages to native or denatured collagen type I and III selectively stimulated the expression of interstitial collagenase and TIMPs, suggesting that ECM components can directly influence macrophage-mediated MMP secretion [52]. Monocytes isolated from the blood of healthy individuals expressed detectable levels of several MMPs including MMP-1, MMP-2, MMP-3, MMP-9, MMP-10, MMP-19, MT1-MMP, MT4-MMP and MT6-MMP under basal conditions [50]. Besides, when granulocyte-macrophage colony-stimulating factor is added in combination with either TNF-α or IL-1β-induced MMP-1 synthesis it synergistically enhanced MMP-9 and TIMP-1 expression. Whereas MMP-9 production is negatively regulated by IL-4, IL-10, IFN-β, IFN-γ and TGF-β in monocytes [50]. Moreover, in vitro studies demonstrate increased expression and activity of both MMP-2 and MMP-9 during monocyte differentiation into macrophages, which emphasises the importance of the cellular state of differentiation in monocyte/macrophage lineage [53]. Similarly, MMP-12 expression in human peripheral blood-derived macrophages is induced by several cytokines and growth factors including IL-1β, TNF-α, M-CSF, VEGF and PDGF-BB [54].
Leukocytes and lymphocytes
Numerous studies have identified a distinctive pattern of MMP expression in different subsets of circulating leukocytes and monocytes under both basal and stimulated conditions [55]. A prominent expression of MMP-11, MMP-26 and MMP-27 in B- cells, MMP-15, MMP-16, MMP-24 and MMP-28 in T-cells, and MMP-7, MMP-8, MMP-21 and MMP-23 expression was found in all leukocyte subsets isolated from healthy individuals [55]. However, a majority of MMPs including MMP-1, MMP-2, MMP-3, MMP-9, MMP-10, MMP-14, MMP-17, MMP-19 and MMP-25 were significantly represented in monocytes. This distinct pattern of MMP expression in monocytes seems to provide an increased transmigratory capacity across a model of the blood-brain barrier, compared with B- and T-cells [55]. In addition, T-cell extravasation into the perivascular tissue during inflammation is mediated by induction and surface localisation of MMP-2 in T-cells via interaction with vascular cell adhesion molecule-1 on ECs [56].
Neutrophils
Neutrophils are potential cellular sources of a wide array of proteolytic enzymes and participate in different inflammatory process by releasing enzymatically active neutrophil elastase, and other proteases including cathepsin G, proteinase 3, MMP-1, MMP-8, MMP-9 and MMP-12 [57]. In addition to the innate immune functions of neutrophils, several studies indicate the role of neutrophil-derived proteases in trans-endothelial neutrophil migration [58] and in the initiation of the angiogenic switch [59]. Specifically, neutrophil-derived MMP-9 is shown to play an important role in basal lamina type IV collagen degradation and in catalysing the angiogenic switch by facilitating MMP-9-dependent VEGF mobilisation and consequent pro-angiogenic signalling [59].
Mast cells
Mast cells release several factors including MMPs, neutral and serine proteinases, heparin, heparinase, tryptase, chymase, histamine, and angiogenic growth factors such as basic fibroblast growth factor (bFGF), VEGF and cytokines [60]. Mast cell-derived neutral proteases like tryptase and chymase potentially mediate activation of latent MMPs in human carotid arteries and subsequently enhance angiogenic phenotypes and also degrade different components of BM [61]. Moreover, in vitro studies show that profibrotic cytokine TGF-β attenuates kit ligand-mediated induction of MMP-9 expression in resident tissue mast cells [62]. Specifically, analysis of the pulmonary artery trunk sections revealed localisation of mast cells in the adventitial layer and diffuse localisation pattern of immunoreactive mast cell-derived collagenase in the media and adventitia of muscular pulmonary arteries [63].
PHYSIOLOGICAL FUNCTIONS OF MMPs IN VASCULATURE
Besides matrix proteolysis in vasculature, MMPs play a crucial role in multiple physiological process like morphogenesis, angiogenesis, tissue remodelling and tissue repair, as well as modulating fundamental cellular processes like proliferation, migration, differentiation, apoptosis, permeability, host defence, release of ECM-bound chemotactic factors and chemokine processing [27, 64].
Angiogenesis
Physiological angiogenesis is a spatially and temporally orchestrated multistep process, which involves the interplay of multiple growth factors that modulate the complex cell–cell and cell–matrix interactions through activation of different proteolytic systems in the vascular microenvironment. In response to an angiogenic stimulus, the endothelial-derived MMPs mediate proteolytic degradation of endothelial cell–cell and EC–matrix interactions and facilitate ECs to acquire a proliferative and migratory or invasive phenotype [65]. In particular, angiogenic factor-induced secretion of MMP-2, MMP-9 and MT1-MMP in vascular ECs is critical for EC proliferation, migration and vascular ECM invasion associated with sprouting angiogenesis [65]. Specifically, gelatinases, MMP-2 and MMP-9 play a critical role in hydrolysing native type IV collagen, a major component of BM to facilitate endothelial sprouting [66]. Furthermore, activation of the focal proteolysis consequently liberates ECM-sequestered pro-angiogenic factors, exposes cryptic pro-angiogenic integrin binding sites in the vascular ECM and also generates pro-angiogenic fragments, which collectively trigger the angiogenic switch [67–69].
Studies with gelatinase- and MT1-MMP-knockout mice provided compelling evidence of the involvement of MMPs in pathophysiological angiogenesis. MMP-2 deficient mice displayed reduced rates of tumour neovascularisation, total vascular area, number of vessels and tumour progression, as well as normal embryonic development of the vascular system [70]. However, the combined deficiency of MMP-2/MMP-9 in the experimental model of tumour angiogenesis displayed an impaired angiogenic and invasive phenotype with strong reduction of gelatinolytic activity; however, the angiogenic and invasive phenotype was not affected by the single deficiency of host MMP-2, MMP-3 or MMP-9 [71]. These studies on double-deficient mice shed light on the in vivo significance of gelatinases and their concerted effect in neovascularisation. Similarly, FGF-2-induced angiogenic response was lacking in the MT1-MMP deficient mice, suggesting that MT1-MMP might be important for initiation of angiogenesis [72]. Furthermore, studies with MMP inhibitors revealed strong inhibition of angiogenic responses both in vitro and in vivo [28].
Cell proliferation and migration
It is well documented that MMPs can directly and indirectly regulate proliferation, migration, invasion and apoptosis of vascular ECs [73] and SMCs [74] through in vitro and inhibitor studies. MMP-2 and MMP-9 in synergy with MT1-MMPs were shown to promote EC cell migration and tube formation by proteolytic remodelling of the BM, by executing focal and controlled dissolution of ECM and by releasing ECM-bound chemotactic factors from the ECM [75, 76]. During neovessel formation, at the leading edge of the developing neovessel, EC proliferation, along with MT1-MMP-dependent activation of MMP-2 and MT1-MMP-dependent focal collagenolysis, was observed [77].
Intimal hyperplasia after arterial injury importantly involves SMC proliferation, migration and ECM remodelling, which may be regulated by various cytokines and growth factors. PDGF-BB or -AB (heterodimer of PDGF subunit A and B/alpha and beta polypeptide)-induced migration of SMCs through medial explants was mediated by MMP-2, whereas the stimulatory effect of bFGF on medial SMC migration was mediated by both MMP-2 and MMP-9 [78]. Administration of potent MMP inhibitor (BB94) dose-dependently suppressed the PDGF-BB-induced migration of cultured SMCs and also suppressed the intimal thickening and medial SMC proliferation after arterial injury [74]. Zymographic studies on MMP expression in the vascular walls revealed constitutive expression of MMP-2 and MMP-9 in normal carotid arteries, while the activated forms were observed in balloon-injured carotid arteries during the phase of SMC proliferation and migration [74]. Moreover, in situ and in vitro studies performed on pulmonary arteries and pulmonary artery smooth muscle cells (PASMCs) from patients showed increased MMP expression, suggesting a critical role for MMPs in different vascular remodelling processes that involves SMC proliferation, migration and intimal thickening [74, 79].
Cell differentiation
Vascular SMCs exhibit remarkable phenotypic plasticity and can be modulated from a quiescent, contractile phenotype to a synthetic phenotype during physiological or pathological vascular remodelling. The proliferative or migratory phenotype of SMC is associated with increased proteolytic activity and ECM turnover, accompanied by loss of differentiation markers including myosin bundles and α-actin composition characteristic of synthetic SMC phenotype [80].
During PDGF-BB mediated recruitment of SMCs and pericytes to neovascular sprouts; vascular SMC dedifferentiate from a contractile to a migratory phenotype, a process shown to be associated with MMP-2 upregulation [81, 82]. Moreover, a study from Risinger et al. [48] demonstrated that the PDGF-BB-induced upregulation of MMP-2 in SMC is antagonised by autocrine expression and activation of TGF-β1 resulting in a significant delay in SMC phenotype switching. In addition, another study identified that TGF-β1 treatment downregulated MMP-1 and MMP-3 production and upregulated TIMP-1 mRNA levels in human myometrial SMCs [83]. Altogether, these findings confirm that TGF-β promotes a contractile phenotype, partly by modulating MMP/TIMP ratios. However, the increased activity of gelatinases, MMP-2 and MMP-9, was shown to be required for transition of differentiated SMC to a synthetic phenotype [84]. Furthermore, elastin peptides generated by serine elastases and/or MMPs stimulate the production of the matrix glycoprotein fibronectin, which changes SMCs from a contractile to a migratory phenotype. With regard to fibroblasts, the homeostatic relationship between resident fibroblasts and the collagen matrix keeps them in a quiescent, undifferentiated state [3]. However, normal fibroblasts subjected to hypoxia display hypoxia-induced phenotypic switching to myofibroblasts, through the MMP-2/TIMP mediated pathway [44]. Specifically, PDGF, TGF-β1, tenascin-C (TN-C), fibronectin and ET-1 exhibit mitogenic activity and induce the myofibroblast phenotype both in vitro and in vivo [85], probably by regulating the MMP/TIMP balance.
Vascular permeability and cell adhesion
Endothelial barrier integrity, vascular permeability and quiescent EC phenotype, which are sustained through endothelial cell–matrix and cell–cell interactions, can be regulated by pro-angiogenic mediators such as VEGF and angiopoietins [73]. Specifically, VEGF promotes vascular permeability by uncoupling inter-endothelial junctions, inducing the formation of endothelial fenestrae and an increase in modification of caveolae [73]. A recent study revealed that the hypoxia-induced vascular leakage in vivo and the associated rearrangement of tight junction protein occludin and its diminished expression is mediated by hypoxia-induced MMP-9 activation. However, VEGF inhibition attenuated vascular leakage, hypoxia-induced MMP-9 activation and dependent gelatinolytic activity. Also, MMP inhibition attenuated vascular hyperpermeability, and prevented gap formation and tight junction rearrangement [74]. Importantly, the integrin, cadherin, selectin and immunoglobin superfamily of endothelial cell adhesion molecules have been identified to play a crucial role in cell–cell and cell–matrix interactions and in different pathophysiological events in vasculature. In particular, the αvβ3 and αvβ5 integrins are described to regulate endothelial cell adhesion and migration during angiogenesis and vasculogenesis [75]. In addition, αvβ3 integrins interact with an array of ECM ligands such as vitronectin, fibronectin, collagen, laminin, von Willebrand factor, fibrinogen, osteopontin, thrombospondin and RGD-containing peptides, which are potential MMP substrates (table S1) [75]. Moreover, during endothelial sprouting, surface expression and MT1-MMP dependent activation of MMP-2 in ECs were found to be associated with integrin αvβ3 and TIMPs at basolateral focal contacts and mediate focal degradation of ECM [76].
Like integrins, cadherins and MMPs are known to a play vital role in determining tissue cohesion, collective cell migration and reorganisation of the ECM during invasion [77]. Studies on in vitro invasion patterns suggest that tissue cohesion correlates with E-cadherin expression. However, the cell lines with intermediate tissue cohesion and relatively high MMP expression exhibit complex migratory patterns [78]. MMP inhibition studies confirm that the decrease in the expression of E-cadherin and increase in type IV collagenase activity (MMP-2 and MMP-9) would enhance detachment of tumour cells and facilitate invasion [79]. In contrast, in ECs, an increase in VE-cadherin was observed in connection with the downregulated MMP production and pericellular proteolysis during stabilisation and maturation of the neovessel [86]. This recent study confirms the mechanism of cell–cell contact dependent regulation of pericellular proteolysis in angiogenesis. This correlates with the finding that MMP inhibitor treatment enhanced colocalisation of cadherin/β-catenin at cell–cell contacts and promoted stabilisation of cadherin-mediated cell–cell adhesion in fibroblasts suggesting a possible feedback loop between MMP and cadherin/β-catenin systems [81].
Mobilisation of growth factors and cytokine processing
Besides the fact that the MMP-mediated breakdown of the ECM barrier facilitates cell migration and invasion, MMP plays a central role in liberation of ECM-bound mitogens through direct digestion of matrix components, regulation of the formation of matrikines and release of biologically active growth factors from the ECM [87, 88]. Besides offering structural support to the vascular tissue, the ECM serves as a repository for bioactive molecules including pro-angiogenic growth factors such as VEGF, bFGF, hepatocyte growth factor, insulin-like growth factor-1, TGF-β1 and connective tissue growth factor (CTGF) [26, 69]. MMP-dependent proteolysis promotes the release of these growth factors from the vascular ECM thus influencing cell proliferation and cell migration [68]. For instance, MMPs regulate the bioavailability of VEGF. VEGF is sequestered as an inactive form by the ECM components like CTGF, pleiotrophin, etc., resulting in reduced bioavailability. Several MMPs (MMP-1, -2, -7, -9, -16 and -19) mediate proteolytic cleavage of this inhibitory complex and increase the bioavailability of VEGF, which plays a key role in various pathophysiological processes [88]. MMP-9 and, to a lesser extent MMP-2, effectively degrade heparan-sulfate proteoglycans or perlecans and increase the mobilisation of the BM-sequestered VEGF [89]. Mice with targeted disruption of MMP-9 exhibited reduced hypertrophic cartilage vascularisation due to the lack of mobilisation of ECM-bound VEGF, confirming the essential role of MMP-9 in bioavailability of growth factors [90]. In addition, in cultured SMCs, serine elastase (EVE) was shown to mediate degradation of elastin, which subsequently promotes liberation of ECM-bound bFGF.
However, MMP-dependent proteolysis of ECM components releases matrikines. Matrikines are fragmented matrix peptides that have biological activities in regulating connective tissue cell activity. For example, MMP-mediated proteolysis of collagen and perlecan generates anti-angiogenic matrikines such as arrestin, canstatin, tumstatin, metastatin, endostatin, neostatin, vastatin, restin and endorepellin [69]. However, MMP-based proteolysis of fibronectin-, laminin-, osteonectin- and elastin-derived matrikines promotes cell proliferation, migration and angiogenesis [88].
Importantly, MMPs also play a crucial role in cytokine processing. TGF-β, a multifunctional cytokine, plays a pivotal role in regulating diverse cellular processes such as proliferation, differentiation, migration, survival and ECM synthesis of vascular cells [91]. TGF-β isoforms are secreted and maintained in a latent form by binding to the elastic microfibrils and ECM. The bioavailability of active TGF-β is mediated by the degradation of microfibrils and the release of the active TGF-β from, latent associated protein-β1 by different proteolytic enzymes like MMP-9, MT1-MMP and plasmin [91]. Besides modulation of cytokine activity and bioavailability, MMPs can mediate chemokine processing and can alter the chemotactic properties of chemokines, consequently influencing the inflammatory response. McQuibban et al. [92] identified that the MMP-2 mediated processing of CCL7/MCP-3 generated a cleaved MCP-3 that acts as a general chemokine antagonist and attenuates chemotaxis and the host inflammatory response in vivo.
DYSREGULATION OF MMPs AND THEIR CONTRIBUTION TO PAH PATHOGENESIS
MMP expression in PAH
Several lines of evidence show disturbed balance of MMPs and TIMPs in the pathogenesis of PH and other lung diseases. As mentioned previously, MMPs contribute to several pathophysiological processes such as ECM turnover, phenotype switching, hyperplasia, cell migration and apoptosis, implicating a crucial role of MMPs in pulmonary vascular remodelling and the establishment of PAH (fig. 2).
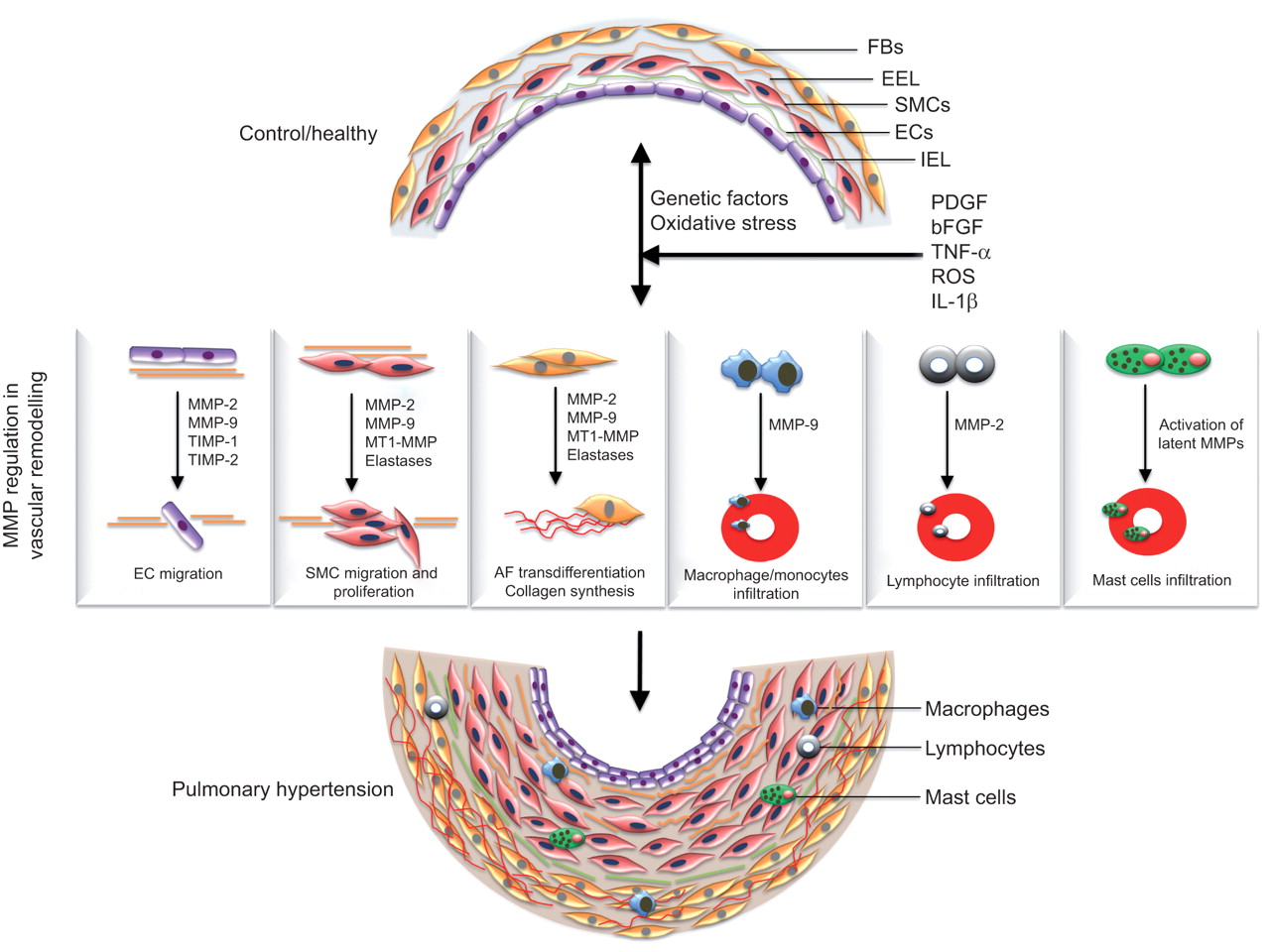
{kind=link}
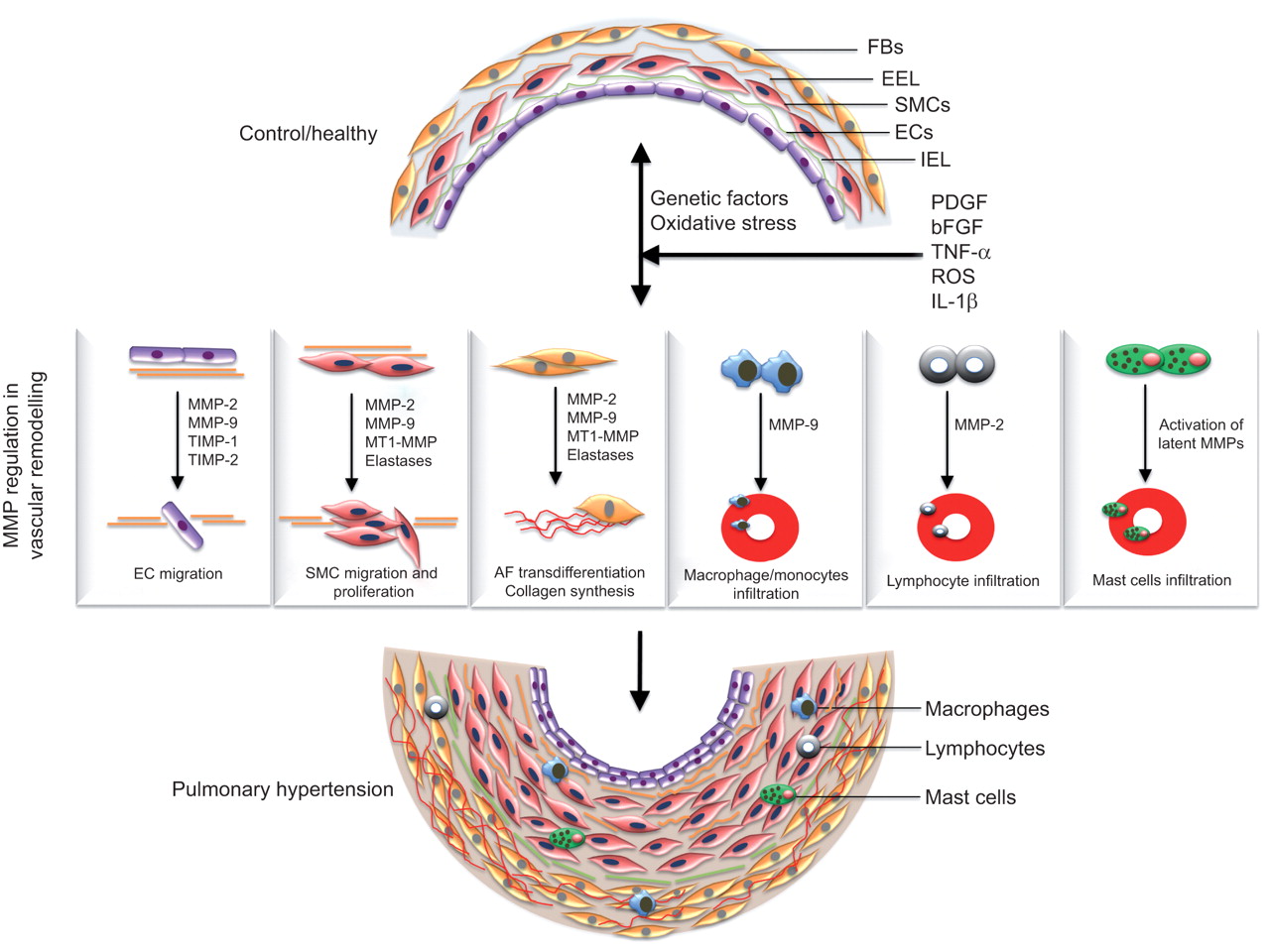{kind=link}
Matrix metalloproteinases (MMPs) in the pathogenesis of pulmonary arterial hypertension (PAH). A variety of physiologic, acquired, and/or exogenous stimuli such as hypoxia, oxidative stress, inflammation, infection and genetic factors cause vascular injury and subsequently promote abnormal production of growth factors and cytokines (such as platelet-derived growth factor (PDGF), epidermal growth factor, fibroblast growth factor-2, transforming growth factor-β, cytokines such as interleukin (IL)-1β, and tumour necrosis factor (TNF)-α) which arbitrate the pathophysiological alterations in vasculature by modulating MMP expression and regulation. Deregulation of MMPs is implicated in several pathophysiological processes such as endothelial cell (EC) migration, smooth muscle cell (SMC) migration and hyperplasia, adventitial fibroblast (AF) trans-differentiation, increased extracellular matrix turnover and recruitment of inflammatory cells, thus facilitating the progression of the pulmonary vascular remodelling process and the establishment of PAH. FB: ; EEL: external elastic lamina; IEL: internal elastic lamina; bFGF: basic fibroblast growth factor; ROS: reactive oxygen species; TIMP: tissue inhibitor of metalloproteinase; MT1: membrane type 1.
Several studies have investigated the expression profile of MMPs (table 1) and serine elastases and their potential role in human PAH. Matsui et al. [98] evaluated the expression and localisation of MMPs in different vascular lesions of PAH through immunohistochemical and immunofluorescence studies. This study found a diffuse and strong reaction for MT1-MMP localised to myofibroblasts and endothelial cells in onion-skin lesions and cellular plexiform lesions. While only a small number of myofibroblasts in both these lesions were positive for MMP-3 and MMP-7. Furthermore, a co-localisation of MT1-MMP and MMP-2 was detected in myofibroblasts and endothelial cells in the cellular plexiform lesions [98]. In particular, discontinuous type IV collagen and focal thinning was observed in onion-skin lesions and mature plexiform lesions. This study emphasises that modulation of MMPs in vascular cells might orchestrate the critical pathological events involved in the development of vascular lesions in PAH [98]. Notably, Lepetit et al. [79] identified increased MMP-2 expression and activity in PASMCs from idiopathic PAH patients. Co-localisation of MMP-2 and gelatinolytic activity in the medial layer along the inner elastic lamina up to the lamina break in idiopathic PAH arteries compared to control specimens was observed [79]. In addition, a significant decrease in MMP-3 and increase in TIMP-1 production was detected in idiopathic PAH PASMCs, favouring ECM accumulation in idiopathic PAH [79]. Although this study was specifically focused on PASMC-derived MMP expression and activity, it confirmed the existence of MMP/TIMP imbalance and increased gelatinolytic activity in the human PH scenario.
Moreover, measurement of circulating MMP levels in a cross-sectional and longitudinal study of hypertensive subjects revealed significantly elevated plasma concentrations of MMP-9 and TIMP-1 at baseline than in the normotensive controls. This significant increase in circulating MMP-9 at baseline in patients with PH could reflect higher vascular and cardiac tissue levels of MMP-9 and increased ECM turnover [96].
MMP expression was mainly studied in two widely employed models of PH: chronic hypoxia-induced PH and monocrotaline (MCT)-induced PH in rodents. In chronic hypoxia-induced PH, exposure of animals to normoxia or hypobaric hypoxia for 2–3 weeks typically leads to 50% increase in mean Ppa and right ventricular hypertrophy. In an MCT-induced PH model, a single subcutaneous injection of MCT (a pyrrolizidine alkaloid of plant origin) after 3–4 weeks causes severe vascular remodelling, chronic PH and cor pulmonale [3, 99].
Microarray and gene expression analysis of MCT-treated lungs revealed differential expression of several genes implicated in ECM regulation and cell adhesion. Upregulation of different MMP subtypes including gelatinases (MMP-2 and MMP-9), neutrophil collagenase (MMP-8), stromelysins (MMP-10, MMP-11), macrophage metalloelastase (MMP-12), MMP-20, PLAT (tPA) and SERPINB2 were identified and validated, demonstrating an indispensable role of MMPs in the establishment of experimental PH. In addition, this study suggests the association of MMPs to enhanced migratory response of ex vivo cultured SMCs derived from MCT-PH arteries [100]. In accordance, significant upregulation of MMP-2, MMP-9 and TN-C as well as increased gelatinolytic activity in isolated pulmonary arteries from MCT-treated animals was confirmed [101]. Moreover, transgenic expression of gelatinase B (human MMP-9) resulted in extensive infiltration of macrophages and ultimately aggravated MCT-induced PH, substantiating the pathological significance of increased MMP-9 in PH [102]. Interestingly, a recent study demonstrated that macrophage-specific transgenic expression of human MMP-1 in a mouse model of MCT-induced PH resulted in attenuation of collagen deposition, SMC proliferation, infiltration of macrophages and consequently medial thickening [103].
Different studies have reported the deregulated expression of vascular MMPs in both classical rodent models of MCT- and hypoxia-induced PH (table 2). Frisdal et al. [110] evaluated expression, activity and localisation of gelatinases specifically in pulmonary vessels during progressive PAH in both experimental models of hypoxia- and MCT-induced PH. Both rodent models of PH revealed strong MMP-2 expression throughout the pulmonary vasculature compared with controls. Importantly, increased gelatinolytic activity was mainly observed in the medial layer and correlates with increased MMP-2 expression in MCT-induced PH pulmonary arteries [110]. However, diffuse distribution of gelatinolytic activity was found in hypoxia-induced PH. Increased gelatinolytic activity can be attributed to MMP-2, due to the absence of MMP-9 expression in both models of PH. Importantly, a correlation between time-dependent increase in MMP-2 gelatinolytic activity and progression of hypoxic PH was observed [110]. Increased collagenolytic activity was also observed in hypoxia-induced PH. Herget et al. [113] confirmed the appearance of collagen breakdown products and increased collagenolytic activity due to hypoxia-induced expression of MMP-13 in pulmonary arteries of hypoxic rats. These experimental studies collectively suggest an important role for MMPs during ECM turnover and pathological vascular remodelling in PH.
Contrary to the findings above that implicate increased MMP activity to severity of PH, some studies have indicated an essential role of MMPs during reverse remodelling process. Thakker-Varia et al. [114] reported a transient increase in the expression of interstitial collagenase (MMP-1), stromelysin-1 (MMP-3) and gelatinases during normoxic recovery from hypoxia-induced PH. A significant increase in stromelysins and total proteolytic, collagenolytic and gelatinolytic activities was predominantly found in the media and adventitia of pulmonary arteries during normoxic recovery of hypertensive vessels [114]. This observed increase in protease activity correlated with the rapid reduction in collagen and elastin content in pulmonary arteries, thus indicating a correlation between decreased vascular remodelling and increased MMP activity during early reversal of hypoxia-induced PH [114, 119]. In a similar study by Tozzi et al. [63], increased activation of mast cell-derived interstitial collagenase was shown to mediate restoration of vascular architecture by facilitating collagen breakdown in remodelled pulmonary arteries during the early recovery phase from chronic hypoxia. Poiani et al. [119] also reported accumulation of collagen and elastin in the main pulmonary arteries of rats during chronic hypoxia, while the normoxic recovery of hypertensive vessels were associated with decreased collagen and elastin content. Cumulatively, these experimental studies suggest an essential role of MMPs in reabsorption of vascular collagen during the de-remodelling process that occurs in the post-hypoxic recovery phase [63, 114, 119].
In addition to MMPs, serine elastases were shown to be dysregulated during PAH pathogenesis. Experimental models of PH showed both an early increase in elastinolytic activity preceding the development of vascular changes and a later increase associated with disease progression. A heightened serine elastases activity and fragmentation of elastin was observed in pulmonary arteries of MCT-induced PAH rats. The increased activity of serine elastase, specifically endogenous vascular elastase (EVE), preceded the development of MCT-induced PAH and the accompanying vascular lesions [120]. In hypoxia-induced PAH, a transient increase in elastase activity was observed [121]. Recently, a transgenic mouse model overexpressing S100A4 developed neointimal lesions after injection of the murine gamma herpes virus 68 (MHV-68) and showed heightened serine elastase activity and elastin degradation after infection of the virus and with reactivation [122].
Consequences of MMP dysregulation in PAH
Abnormal production of ECM components
Homeostasis of the medial ECM components like elastin, vascular collagen, fibronectin, TN-C and their turnover by MMPs plays a vital role in regulating the phenotype switching of SMC and associated PASMC hypertrophy, hyperplasia, migration and medial ECM turnover [123]. Increased deposition of collagen and elastin are known to be important determinants of medial thickening during the progression of PAH [123]. In addition, evidences also shows increased accumulation of collagen and elastin in the vessel wall as one of the vital contributing factors to the progression of chronic hypoxia-induced PH [119]. Collagen accumulation contributes to pulmonary arterial stiffening, which is implicated in PH progression and right ventricular dysfunction through two mechanisms: increased distal arterial cyclic strain damage, which promotes SMC proliferation; and proximal wave reflections, which increase right ventricular afterload [124].
Ultrastructural assessment of pulmonary arteries in lung biopsy tissue from patients with PAH showed fragmentation of the IEL [125]. Furthermore, gaps in the IEL were also reported in patients with PAH and neointimal lesions [126]. In agreement with this observation, animal studies showed fragmentation of elastin preceding the development of vascular changes. Elastin peptides generated by serine elastases and/or MMPs stimulate the production of the matrix glycoprotein fibronectin, which changes SMCs from a contractile to a migratory phenotype [123].
Another consequence of MMP and elastase activation is the increased production of glycoprotein TN-C in PASMCs that positively regulates PASMC proliferation. Increased expression of the TN-C is associated with progression of clinical and experimental PH [123]. In support, treatment of organ cultures with either MMP-2 or an elastase inhibitor resulted in suppression of TN-C expression, and regression of medial hypertrophy associated with PASMC apoptosis [111]. Subsequent studies in cultured PASMCs documented that TN-C amplifies the mitogenic response to FGF-2 and is a prerequisite for EGF-dependent SMC proliferation [127].
Regulation of key molecules in PAH
Several growth factors (such as PDGF, EGF, FGF-2, bone morphogenetic protein (BMP) and TGF-β), vasoactive substance such as endothelin, cytokines such as IL-1β, and TNF-α, serotonin and ROS are demonstrated to be deregulated in PAH [128]. These factors arbitrate pathophysiological alterations such as hyperplasia, hypertrophy, migration, phenotypic modulation of vascular cells and consequently pulmonary vascular remodelling [7, 80, 128]. Importantly, a potential crosstalk between these pro-hypertensive molecules and MMPs/serine elastases has been observed. Knockdown of BMPR1A in human PASMCs reduced MMP-2 and MMP-9 activity, attenuated serum-induced proliferation, and impaired PDGF-BB-directed migration. Furthermore, knockdown of MMP-2 or MMP-9 recapitulated these abnormalities, supporting a functional interaction between BMP signalling and MMPs [129]. A direct association between MMPs and growth factor receptors has been suggested. Serum induction of PASMC elastase was shown to signal through the activation of tyrosine kinase [130]. However, serine elastases can degrade the ECM and consequently release SMC growth factors such as bFGF. This function could be amplified via activation of MMPs [38]. MMPs also positively regulate EGF-dependent SMC proliferation by promoting TN-C induced clustering of αvβ3 integrin receptor [111]. In addition, studies from genetic or pharmacological ablation of serotonin receptor 5-HT2BR suggests that 5-HT2BR-dependent increased elastase activity and MMP/TIMP imbalance, subsequently leading to latent growth factor release, including TGF-β [105, 131]. Vice versa, evidence suggests that TGF-β1 stimulates expression of pro-MMP-9 in IL-1β treated PASMCs. IL-1β, a major cytokine associated with PH, was shown to markedly increase MMP-2 and MMP-9 gelatinase activity [132]. With regard to endothelin system, elevated MMP-9 activity was found in endothelin-B receptor-deficient rats with PH, suggesting endothelin-mediated regulation of MMPs [117].
THERAPEUTIC USE OF MMP INHIBITORS IN EXPERIMENTAL MODELS OF PH
Several pharmacological studies of MMP inhibition were mostly performed in two of the experimental models of PH: hypoxia- and MCT- induced PH (table 3). Cowan et al. [111] reported the therapeutic efficacy of elastase and MMP inhibitors in the MCT-induced PH model. This effect is associated with reduced TN-C, suppression of SMC proliferation and induction of apoptosis. Moreover, selective repression of TN-C (a matrix molecule induced by MMPs) by transfecting pulmonary arteries with antisense/ribozyme constructs also induces SMC apoptosis and arrests progressive vascular thickening but fails to induce regression of the disease. Vieillard-Baron et al. [6] demonstrated that intratracheal instillation of the adenovirus-mediated overexpression of human TIMP-1 gene in the lungs of rats exposed to MCT reduced pulmonary vascular remodelling, right ventricular hypertrophy, gelatinase activity and muscularisation of peripheral pulmonary arteries, suggesting that balancing the MMP/TIMP ratio can reverse the disease.
However, mixed results were obtained with MMP inhibition in the hypoxia-induced PH model. Herget et al. [113] demonstrated that chronic hypoxia induces expression of rat interstitial collagenase (MMP-13) in peripheral pulmonary arteries and, thus, treatment with synthetic MMP inhibitor batimastat diminished muscularisation of distal lung vessels and markedly attenuated hypoxia-induced PH. This study emphasised that the stimulation of collagenolytic activity is a substantial causative factor in the pathogenesis of hypoxia-induced pulmonary vascular remodelling and hypertension. Furthermore, increased synthesis and accumulation of collagen and elastin was identified in the main pulmonary arteries of rats during early development of hypoxic PH [119]. In accordance, a study by Kerr et al. [135], using inhibitors for collagen synthesis (cis-4-hydroxy-L-proline) and crosslinking (β-aminopropionitrile) in the rats exposed to chronic hypoxia, demonstrated reduction of excess vascular collagen and attenuation of PH. However, in contrast, Vieillard-Baron et al. [134] demonstrated that intratracheal instillation of the adenovirus-mediated overexpression of human TIMP-1 gene or administration of a broad-spectrum MMP blocker (doxycycline) in rats subjected to chronic hypoxia was associated with increased muscularisation and periadventitial collagen accumulation in distal arteries. This study shows that inhibition of lung MMPs in rats subjected to chronic hypoxia ultimately contributes to significant exacerbation of pulmonary artery remodelling and aggravation of PAH.
Apart from MMP inhibition, serine elastase inhibition (M249314 or ZD0892) has shown to attenuate [136] or reverse MCT-induced PH [136]. Initially, in ex vivo organ culture studies using MCT-induced PH rat pulmonary arteries have shown that elastase inhibitors suppress TN-C and induce SMC apoptosis [111]. This initiates complete regression of the hypertrophied vessel wall by a coordinated loss of cellularity and ECM. Accordingly, in vivo elastase inhibitors were shown to reverse advanced pulmonary vascular disease in MCT-induced PH rats [137]. Similarly, administration of a serine elastases inhibitor (SC-39026) to rats reduced hypoxia-induced PH [121]. Furthermore, transgenic mice that overexpress the serine elastases inhibitor elafin when exposed to chronic hypoxia demonstrated reduced serine elastase and MMP activity compared to the non-transgenic mice. Importantly, elafin-transgenic mice displayed reduced right ventricular pressure, reduced muscularisation and preservation of the number of distal vessels as compared with control or non-transgenic mice [40].
Furthermore, apart from MMP inhibitors, several pharmacological compounds regressing established PH seem to function via modulation of MMP/TIMPs. Pullamsetti et al. [100] demonstrated that inhalation of a combined selective PDE3/4 inhibitor (tolafentrine) exhibited anti-proliferative, anti-migratory and anti-remodelling effects, and consequently reversed MCT-induced PH in rats. This study showed that the reverse remodelling effects of tolafentrine is due to downregulation or even normalisation of the deregulated profile of several MMPs and adhesion molecules in response to MCT. Lercanidipine, a vasoselective dihydropyridine calcium channel blocker, demonstrated beneficial effects in patients with PH by decreasing elevated circulating MMP-9 levels [93, 96]. This lercanidipine-induced effect was associated with a significant decrease in MMP-9 activity without affecting proMMP-2 activity and TIMP-1 concentration besides a reduction in oxidative stress in patients with PH and with PH and diabetes mellitus [93]. Similarly, administration of a third-generation calcium channel blocker, amlodipine, immediately followed by MCT treatment suppressed the MCT-induced increase in MMP-2 activity, platelet activation, EC damage and SMC proliferation, and consequently inhibited the development of PH [133]. Administration of the FDA-approved drug for PH, bosentan (dual endothelin receptor agonist, ETA/B), also attenuated the MCT-induced upregulation of MMP-2, TIMP-1, endothelial NO synthase expression and MCT-induced PH [104]. Amelioration of MCT-induced PH by fluoxetine (selective serotonin reuptake inhibitor) treatment was associated with suppression of MMP-2, MMP-9, TIMP-1 and TIMP-2 expression [105, 106]. In a recent study, captopril (an angiotensin-converting enzyme inhibitor) and losartan (angiotensin II type 1 receptor antagonist) administration attenuated pulmonary vascular remodelling, probably associated with the regulation of the expressions of MMP-2, MMP-9 and TIMP-1, in pneumonectomy plus MCT injection-induced severe PAH [115].
The pharmacological studies of MMP inhibition in hypoxia- and MCT-induced experimental PH models have substantiated that selective or pan-MMP inhibition can be bidirectional: attenuates or exacerbates vascular remodelling and PH. The differential outcome could partly suggest the existence of distinctive mechanisms involving ECM accumulation and MMP activity underlying the development of experimental PH. Moreover, clinical studies performed in diseases such as cancer and arthritis have hinted at the possible dose-limiting side-effects of pan-MMP inhibition, such as musculoskeletal syndrome that manifests as pain and immobility in the shoulder joints, arthralgias, contractures in the hands and reduced overall quality of life in patients [138]. Pre-clinical studies using selective MMP inhibitors (table S2) might alleviate the aforementioned issues.
CURRENT ISSUES AND FUTURE PERSPECTIVES
Deregulated expression and activity profiles of MMPs/TIMPs have been detected in human PAH and experimental models of PH. Moreover, several pharmacological studies of MMP inhibition in different experimental animal models of PH have substantiated that selective or pan-MMP inhibition can attenuate or enhance vascular remodelling and PH. Thus, it is still not clear whether inhibition of MMPs represents a useful strategy for the treatment of PAH. Several issues need to be addressed before considering MMP inhibition as a clinical therapeutic strategy.
The conflicting outcomes of MMP inhibition studies in hypoxia and the monocrotaline model of PH in terms of regressing pulmonary vascular lesions suggest that MMP inhibition mediated beneficial effects depend upon the primary insult involved, or the type of inhibitor used. In addition, this can also strongly support the existence of distinctive mechanisms underlying in the development of hypoxia-induced PH as compared with MCT-induced PH and their differential responses to pharmacological agents.
A detailed characterisation of MMP expression and activity in different sub-types of human PH is lacking, especially in pulmonary vasculature and in different cell types of pulmonary vasculature. Extrapolating the animal data to human PH subgroups suggest that the chronic hypoxic models of PH in rodents regarded to have relevance to group 3, i.e. human PH associated with hypoxia (PH associated with pulmonary parenchymal disease, sleep disordered breathing, severe chronic obstructive pulmonary disease and residence at high altitude), demonstrate increased ECM turnover, i.e. increased elastolytic and gelatinolytic activity, as well as accumulation of ECM proteins. However, MCT-induced PH is regarded as group 1, PAH caused by drugs or toxins. In this subgroup increased ECM turnover may play a predominant role. In this context, acquiring more information about the specific MMP/TIMP pattern in different subgroups of human PH is of extreme importance.
Additionally, neither chronic hypoxia- nor MCT-induced animal models of PH perfectly resembles the complex human situation. Both models trigger only mild-to-moderate PH and do not recapitulate neointimal proliferation and plexiform lesions that are important hallmarks of severe PH [139]. This makes it extremely difficult to extrapolate the outcome of MMP inhibition in animal models to humans. Nevertheless, a greater understanding of the involvement of MMPs in experimental models of neointimal lesions or plexogenic lesions is indispensable to further explore the possibilities of therapeutic MMP inhibition in PAH.
Importantly, broad-spectrum MMP inhibitors, such as marimastat, failed to demonstrate clinical efficacy due to severe side-effects. The most frequent side-effect associated with the clinical trials of MMP inhibitors was a musculoskeletal syndrome [138]. Despite this, periostat is the only MMP inhibitor that has been approved by the FDA for the treatment of periodontal disease. Possible reasons for the low success rate of MMP inhibitors in the clinic include unwanted side-effects caused by their lack of selectivity, poor oral bioavailability and decreased potency in vivo. To bypass this it is fundamental to know which MMP in the PH pulmonary vasculature is to be targeted using selective MMP inhibitors and the stage of the disease to allow better selectivity and efficacy. The systemic toxicity could potentially be overcome by aerosol delivery of the drug, permitting local administration and a lower dose of the compound.
Importantly, the lack of selective MMP inhibitors, together with the limited knowledge about the exact functions of a particular MMP, hampers the clinical application. Third generation of MMP inhibitors is currently under investigation and may potentiate reverse remodelling process in PH, without any detrimental off-target effects. However, this has to be screened in appropriate in vitro and in vivo models to evaluate therapeutic benefit.
Previously, most of the in vitro studies are performed by two-dimensional culture of pulmonary vascular cells. However, the in vivo vascular cell matrix interactions are complex and reiterate the necessity to employ three-dimensional culture systems or organ culture models. In addition, it is important to identify the substrates/gene targets and crosstalk between MMPs and other signalling pathways relevant to PH pathogenesis need to be investigated. Clearly, assessing the pre-clinical efficacy of selective MMP inhibitors in severe animal models of PH is warranted. In conclusion, MMPs plays an indispensible role in pulmonary vascular remodelling processes and may be an attractive target for the treatment of PH.
Footnotes
This article has supplementary material accessible from www.erj.ersjournals.com
Previous articles in this series. No. 1: Löffek S, Schilling O, Franzke C-W. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J 2011; 38: 191–208. No. 2: Elkington PT, Ugarte-Gil CA, Friedland JS. Matrix metalloproteinases in tuberculosis. Eur Respir J 2011; 38: 456–464. No. 3: Gaggar A, Hector A, Bratcher PE, et al. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur Respir J 2011; 38: 721–727. No. 4: Davey A, McAuley DF, O’Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J 2011; 38: 959–970. No. 5: Vandenbroucke RE, Dejonckheere R, Libert C. A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur Respir J 2011; 38: 1200–1214. No. 6: Dancer RCA, Wood AM, Thickett DR. Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J 2011; 38: 1461–1467. No. 7: Churg A, Zhou S, Wright JL. Matrix metalloproteinases in COPD. Eur Respir J 2012; 39: 197–209. No. 8: Dagouassat M, Lanone S, Boczkowski J. Interaction of matrix metalloproteinases with pulmonary pollutants. Eur Respir J 2012; 39: 1021–1032.
Statement of Interest
None declared.
- Received November 30, 2011.
- Accepted April 3, 2012.
- ©ERS 2012
REFERENCES











Jump To
- Article
- Abstract
- PULMONARY HYPERTENSION
- ECM OF VASCULATURE
- MATRIX METALLOPROTEASES, ADAMALYSINS, SERINE ELASTASES AND THEIR INHIBITORS
- EXPRESSION AND REGULATION OF MMPs IN VASCULATURE
- PHYSIOLOGICAL FUNCTIONS OF MMPs IN VASCULATURE
- DYSREGULATION OF MMPs AND THEIR CONTRIBUTION TO PAH PATHOGENESIS
- THERAPEUTIC USE OF MMP INHIBITORS IN EXPERIMENTAL MODELS OF PH
- CURRENT ISSUES AND FUTURE PERSPECTIVES
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics