





Abstract
Airway smooth muscle (ASM) is the major effector of excessive airway narrowing in asthma. Changes in some of the mechanical properties of ASM could contribute to excessive narrowing and have not been systematically studied in human ASM from nonasthmatic and asthmatic subjects.
Human ASM strips (eight asthmatic and six nonasthmatic) were studied at in situ length and force was normalised to maximal force induced by electric field stimulation (EFS). Measurements included: passive and active force versus length before and after length adaptation, the force–velocity relationship, maximal shortening and force recovery after length oscillation. Force was converted to stress by dividing by cross-sectional area of muscle.
The only functional differences were that the asthmatic tissue was stiffer at longer lengths (p<0.05) and oscillatory strain reduced isometric force in response to EFS by 19% as opposed to 36% in nonasthmatics (p<0.01).
The mechanical properties of human ASM from asthmatic and nonasthmatic subjects are comparable except for increased passive stiffness and attenuated decline in force generation after an oscillatory perturbation. These data may relate to reduced bronchodilation induced by a deep inspiration in asthmatic subjects.
- Airway hyperresponsiveness
- airway mechanics
- asthma
- force–velocity relationships
- length–tension relationships
- smooth muscle
Asthma is characterised by exaggerated airway narrowing caused by airway smooth muscle (ASM) shortening. However, it is unclear whether there is a fundamental phenotypic change in the ASM itself or if the nonmuscle components of the airway wall or surrounding lung parenchyma are primary contributors to this airway hyperresponsiveness (AHR) [1, 2]. A major hurdle to a clear understanding of ASM contractile function in disease has been the limited data. Of the 12 studies in which ASM mechanical properties have been compared in asthmatic and nonasthmatic tissue, seven have demonstrated no differences [3–9], while five have shown increases in force, shortening or agonist sensitivity [10–14].
We have previously demonstrated that ASM cell bundles carefully dissected from the tracheas of nonasthmatic subjects whose lungs were donated for medical research provide a valuable, high quality tissue preparation for study of the mechanical properties of ASM [15]. We showed that the mechanical properties of nonasthmatic ASM were similar to those measured in other mammalian models. This is in contrast to previous studies which suggested that human ASM produced less force per unit area and shortened less than the ASM of other mammals [16].
The purpose of this study was to re-evaluate a series of hypotheses related to ASM mechanics that have been suggested as possible defects in asthmatic ASM function and potential contributors to AHR. These include determining whether asthmatic ASM produces more stress (force per unit cross-sectional area of muscle) than nonasthmatic ASM [17, 18]; whether the length–tension relationship of asthmatic ASM or its ability to undergo length adaptation are altered [19, 20]; whether the muscle shortens faster or more extensively in asthmatics than nonasthmatics at different loads [21–24]; and whether asthmatic ASM responds differently to a mechanical perturbation than nonasthmatic ASM [25, 26].
We found that passive stiffness was greater and the force reduction following a length perturbation was less in the asthmatic ASM preparations compared with nonasthmatic ASM. These abnormalities could contribute to altered in vivo airway function and increased airway responsiveness.
MATERIALS AND METHODS
Tissue preparation and equilibration
The lungs of 12 asthmatic and nine nonasthmatic subjects were used for these studies. The human lungs were donated for research through the International Institute for the Advancement of Medicine (IIAM, Edison, NJ, USA; www.iiam.org). Donor deaths were primarily because of head trauma in the nonasthmatics, while eight of 12 asthmatics died during exacerbations of their asthma. The subject demographics and clinical details are shown in table 1.
The study was approved by the University of British Columbia–St Paul's Hospital ethics committee (Vancouver, BC, Canada). The preparation of the tissue and details of the mechanical and morphometric measurements were as previously described [15, 27] and are detailed in the online supplementary material. Airway dimensions were measured on intraparenchymal airways, ASM mechanics on trachealis muscle strips. The measurements included: 1) airway dimensions on intraparenchymal airways; 2) maximal trachealis muscle isometric force (Fmax) at in situ length (Lref); 3) passive and active force–length relationships (lengths of 0.5, 0.75, 1.0, 1.25 and 1.5 Lref); 4) force–velocity curves at two time-points (the peak of tetanic force and midway to the peak [15, 28]); 5) maximal isotonic shortening; and 6) force recovery following a 10-min 0.2 Hz oscillation of the relaxed muscle (60% Lref peak-to-peak amplitude, i.e. 30% lengthening; our lever system was not able to apply half sine waves (stretch only)). When the compliance of the lever system was taken into account, the actual stretch applied to the muscle was ∼25%. The maximal stress produced by the trachealis muscle was determined by dividing Fmax (mN) by the cross-sectional area (mm2) of muscle present in the preparation.
Statistical analysis
For the analysis of airway dimensions we compared groups and calculated individual data for comparison with the trachealis muscle physiology. A linear mixed-effects model was used to compare the area measurements of epithelium, lamina propria, smooth muscle, adventitia and total wall, all referenced to basement membrane perimeter (see online supplement for details). A similar analysis was applied to the relationships between the square root of the airway compartment areas and the basement membrane perimeter. Using this relationship the airway smooth muscle area for an airway with a diameter of 1 mm was calculated for each subject for comparison with the physiological results from their trachealis muscle.
Force and length measurements were normalised to Fmax or Lref, respectively. Velocity of shortening was expressed as ΔLref·s−1. Aggregate data were expressed as mean±sem. One- and two-way ANOVA and regression analyses were accomplished using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). p≤0.05 was considered to be sufficient to reject the null hypothesis.
RESULTS
Trachealis muscle mechanics were determined on eight out of 12 and six out of nine of the asthmatic and nonasthmatic subjects, respectively; six of the asthmatics on whom trachealis mechanics were performed had died from an asthmatic attack. For seven samples on which muscle mechanics were attempted, little or no force could be measured in response to electrical field stimulation (EFS). The average ages of the nonasthmatics and asthmatics were 25.3±8.1 yrs and 15.7±2.3 yrs, respectively (unpaired t-test, p=0.247).
Trachealis and intra-parenchymal airway morphology
The mean percentage of ASM in the trachealis preparations of the nonasthmatic subjects was 25.5±9.0% while, in the asthmatics, it was 28.8±8.7% (p=0.53). The mean percentage of connective tissue in muscle bundles in the nonasthmatic subjects was 39.8±5.7% and, in the asthmatics, it was 31.9±11.2% (p=0.12). The airway wall dimensions of 207 airways from 12 asthmatic and nine nonasthmatic donor lungs were analysed (table 2). The ratios of area to basement membrane perimeter (Pbm) for the smooth muscle (p<0.01), lamina propria (p=0.013), adventitia (p=0.020) and total wall (p=0.024) were greater in the asthmatic than the nonasthmatic subjects, while the epithelial area was not significant (p=0.053). A similar analysis showed that the slope of square root of ASM area versus Pbm was steeper (p>0.001) in the asthmatic subjects compared with the nonasthmatic subjects (fig. S2 in online supplementary material). The rationale for using the actual wall areas in the statistical analysis and the square root of the wall area in the graphical analysis is described in the online supplementary material.
Trachealis muscle mechanics
Muscle stress generated with maximal EFS was 152.9±107.7 kPa in the asthmatic and 161.0±50.7 kPa in nonasthmatic preparations (p=0.858) (fig. 1).

Comparison of maximal stress generation. Stress in response to electric field stimulation, calculated as maximal trachealis muscle isometric force divided by muscle cross-sectional area (kPa), between nonasthmatics and asthmatics was compared. Error bars indicate sem. n=6 for nonasthmatics and n=8 for asthmatics.
Force–length properties are shown in figure 2a. Immediately following a length change, the active force produced by the muscle declined and gradually recovered over the 20-min period in which the muscle length was held constant and EFS was applied at 5 min intervals (fig. 2b). After each length change to either a shorter or longer length than Lref, and the determination of immediate force and adapted force at that length, the muscle was returned to Lref to allow for a period of adaptation. Except for the asthmatic group at 1.25 Lref, force did not adapt back to the level of Fmax at any of the length steps. The changes in passive force following length changes and repeated stimulations are shown in figure 2c. The passive force increased dramatically when length was changed to longer lengths and decreased immediately following a change to a shorter length. While the passive force at longer lengths declined over the 20-min period, it remained elevated in both groups when compared with the passive force at Lref.

Time course of length changes and the resulting active and passive force. Airway smooth muscle strips were either shortened or lengthened in the relaxed state to 0.50, 0.75, 1.25 or 1.50 at in situ length (Lref). The muscle was stimulated to contract five times with electric field stimulation (EFS) at 5-min intervals after every length change. Between shortening and lengthening steps, the muscle was returned to Lref and readapted. Grey hexagons indicate the time-points where EFS occurred (a). The b) active and c) passive forces were recorded at every contraction. Both were normalised to maximal trachealis muscle isometric force (Fmax). •: nonasthmatics; •: asthmatics. Error bars indicate sem; n=6 for each group.
The extent of length adaptation and a comparison between groups is illustrated in figure 3. To quantify length adaptation, the first contraction after a length change was compared with the last contraction at that length. For both the nonasthmatics and the asthmatics, significant length adaptation occurred (ANOVA, p<0.01). For the nonasthmatic subjects, Bonferroni post tests revealed significantly greater force following adaptation at 0.5 Lref (p<0.05) and 1.5 Lref (p<0.01) but not at 0.75 and 1.25 Lref. The asthmatics demonstrated greater active force following adaptation at 1.25 and 1.50 Lref (p<0.01) but not at 0.75 or 0.5 Lref. Before adaptation, the active force at 1.50 Lref was significantly lower in asthmatics compared with nonasthmatics (p=0.05) and after the adaptation period the asthmatics produced less force than the adapted nonasthmatics at 0.50 Lref (p<0.01). Figure 4 shows the changes in passive force with changes in length and during the adaptation process. Both the nonasthmatics and asthmatics demonstrated significant passive force adaptation, i.e. the passive force decreased significantly after the period of adaptation in both groups (ANOVA, p=0.024 for nonasthmatics and p=0.018 for asthmatics). Bonferroni post tests demonstrate that the passive force at 1.50 Lref for the asthmatics was significantly greater than that of the nonasthmatics before adaptation (p<0.05). In addition, the passive force at 1.50 Lref was significantly lower in the asthmatics after adaptation than before adaptation (p<0.05).

Active length–force relationship before and after length adaptation. From in situ length (Lref), the airway smooth muscle strips were either shortened or lengthened to 0.50, 0.75, 1.25, and 1.50 Lref (refer to fig. 4 for sequence and time course of length changes and stimulation). Active force before (solid lines and closed symbols) and after (dashed lines and open symbols) length adaptation are shown. Fmax: maximal trachealis muscle isometric force. Error bars indicate sem, n=6 for each group. *: p<0.05, ***: p<0.01; compared with the nonasthmatic before length adaptation at the given length. ###: p<0.01; compared with the asthmatic before length adaptation at the given length. ¶: p<0.05; compared with the nonasthmatic after length adaptation at the given length.

Passive length–force relationship before and after length adaptation. From in situ length (Lref), the airway smooth muscle strips were either shortened or lengthened to 0.50, 0.75, 1.25 and 1.50 Lref. Passive force both before (solid lines and closed symbols) and after (dashed lines and open symbols) length adaptation are shown. Fmax: maximal trachealis muscle isometric force. Error bars indicate sem, n=6 for each group. *: p<0.05, passive force was greater in asthmatics compared with the nonasthmatic before length adaptation at 1.50 Lref. #: p<0.05; passive force was less following adaptation in the asthmatic tissue compared with before adaptation at 1.50 Lref.
Two sets of force–velocity relationships were determined: one during the early phase of contraction and one at the peak of tetanic contraction, as previously described [15, 28]. The force–velocity curves for individual muscle strips varied as shown in supplementary figures S3A and B. Force–velocity data for each muscle strip were initially fitted with Hill's hyperbolic equation [29] before averaging the curves (fig. 5). There were no differences in the shape or position of the force–velocity relationships between nonasthmatic and asthmatic subjects.

a) Averaged isotonic shortening velocities to various loads, early phase release. Hill's hyperbolic equation was fitted to the data from each subject and the solid lines represent the average of these curves. b) Averaged isotonic shortening velocities to various loads, late phase release. Hill's hyperbolic equation was fitted to the data from each subject and the solid lines represent the average of these curves. The dashed lines indicate sem. Lref: in situ length; Fmax: maximal trachealis muscle isometric force.
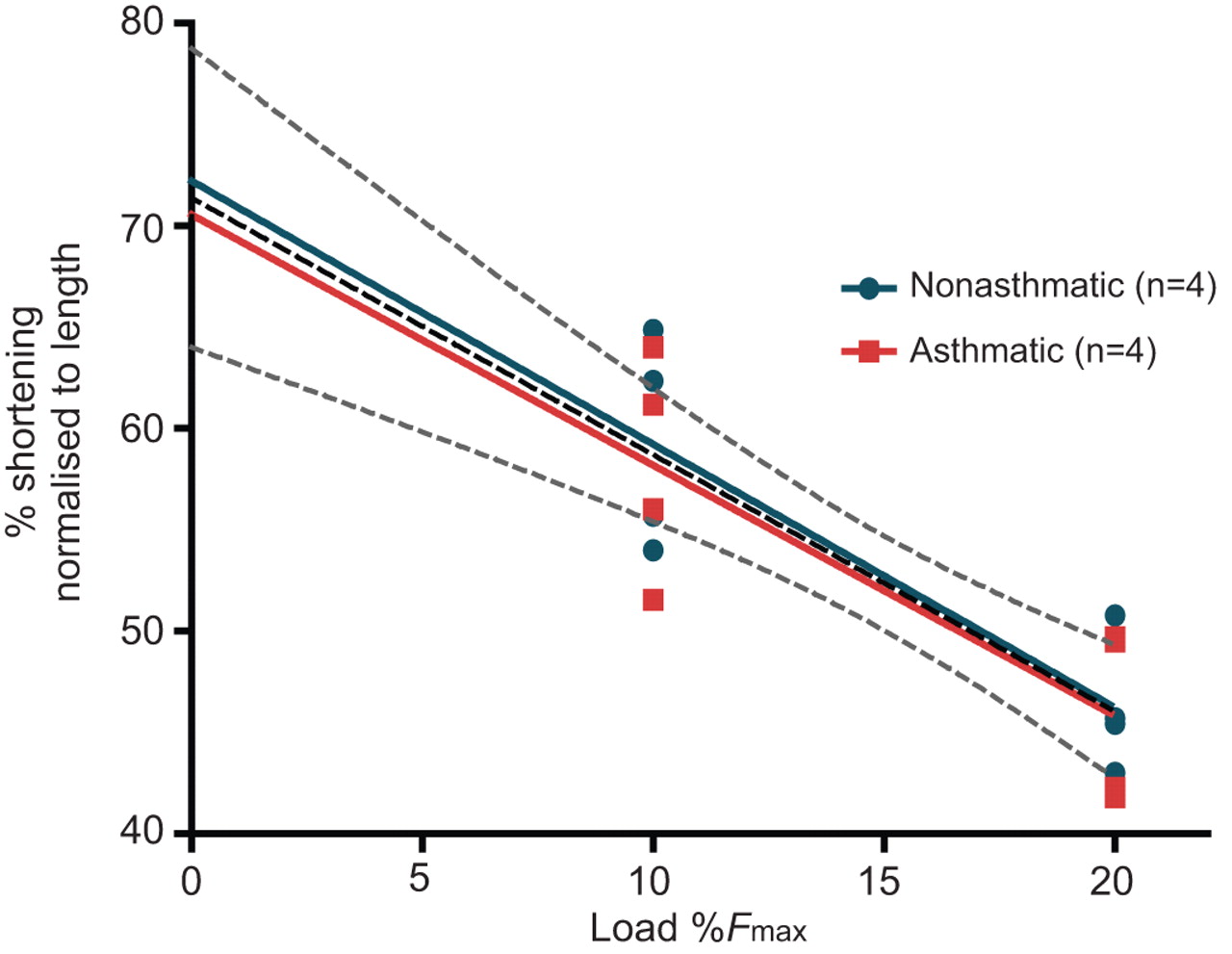
Four nonasthmatic and four asthmatic muscle strips were analysed to determine maximal isotonic shortening (fig. 6). The extent of shortening was recorded at pre-loads of 10% and 20% of EFS-induced Fmax. Maximal shortening, as determined by extrapolation to no load, was 72.2±4.9% for the nonasthmatics and 70.5±5.6% for asthmatics (p=0.946). With no difference between the groups, all muscle strips were combined into one linear regression (central dashed line, fig. 6) and maximal shortening was calculated as 71.4±3.4% (p<0.01, r2=0.708). In the unloaded condition, the 95% confidence intervals were 64.0–78.8% shortening.
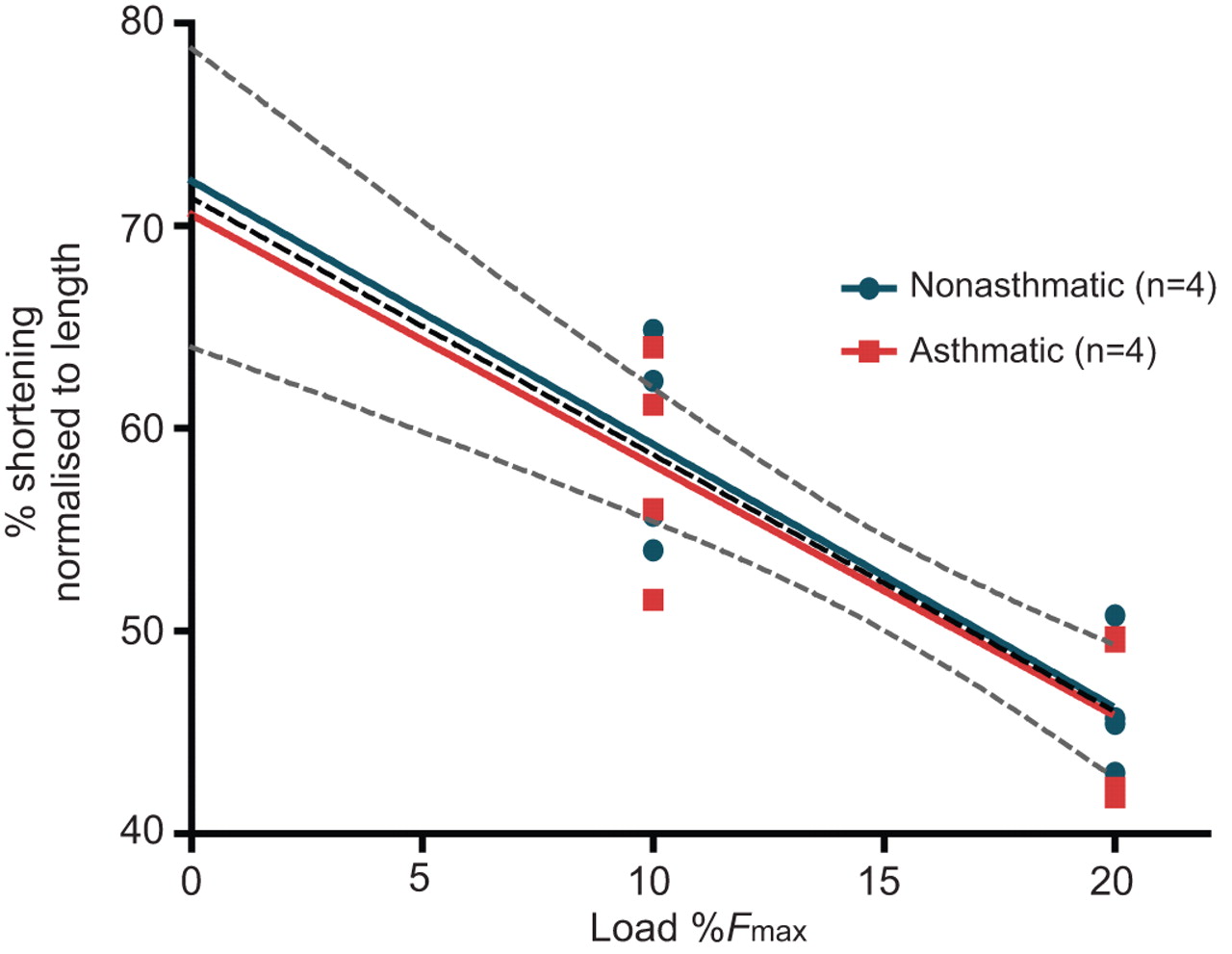
Maximal isotonic shortening. Airway smooth muscle strips were stimulated to contract with electric field stimulation and the total amount of isotonic shortening against small loads was measured. Using the amount of shortening recorded at loads representing 10% and 20% of maximal trachealis muscle isometric force (Fmax), maximal shortening was extrapolated. Shortening was recorded at both loads per donor. n=4 for each group. A combined (both groups) linear regression is represented as a dashed central line. The upper and lower dashed lines represents the 95% confidence interval for all data points.
Active force recovery following a 10-min length oscillation was followed for 30 min in both nonasthmatic and asthmatic tracheal strips (fig. 7). The response to oscillation was significantly different between asthmatics and nonasthmatics (p<0.01 for two-way ANOVA with time and group as variables). Initially following oscillation the nonasthmatics produced an average force of 0.63±0.03 Fmax compared with 0.81±0.04 Fmax in asthmatics (Bonferroni post tests, p<0.01). The recovery of force was greatest over the first 5 min in the nonasthmatics; however, the average force recovery was below Fmax at 0.95±0.03 Fmax. The asthmatics demonstrated slower force recovery but recovered beyond Fmax after 30-min (1.05±0.03 Fmax).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Response to mechanical perturbation by plotting the recovery of isometric force following a 10-min, 0.2 Hz, 30% in situ length (Lref) oscillation (grey shading). Airway smooth muscle strips were adapted to Lref prior to oscillation. The force produced by seven electric field stimulation-induced contractions was recorded at 5-min intervals following the oscillation. ANOVA: p<0.01. Error bars indicate sem, n=6 per group. In addition to the difference in the initial decline in force, the asthmatics demonstrated slower force recovery but recovered beyond maximal trachealis muscle isometric force (Fmax) after 30-min (1.05±0.03 Fmax). Nonlinear regression analysis of the nonasthmatic and asthmatic data demonstrated these differences in recovery rate. The regressions were fitted using a one-phase exponential equation. For the nonasthmatics: force=0.947-0.296e-0.139t (r2=0.73); for asthmatics: force =1.053-0.243e-0.073t (r2=0.48), where force was relative to Fmax and t was time in minutes.
As ASM area, normalised to Pbm, was the morphological variable that best separated asthmatic from nonasthmatic airways, we calculated the smooth muscle area at a Pbm of 3,140 μm (which conforms to an airway with a diameter of 1,000 μm) to use as a continuous variable to compare with trachealis muscle function. Although this variable separated asthmatics from nonasthmatics (0.27±0.15 μm2 versus 0.20±0.09 μm2 p=0.07), there were no significant relationships between this variable and any of the functional variables when we included all subjects in the analysis or when we limited the analysis to asthmatics. Figure S4 shows the relationship of trachealis muscle stress versus the ASM area in an idealised airway with a diameter of 1,000 μm. Similar results were obtained with use of the slope of the relationship between the square root of ASM area and Pbm as the morphological estimate of airway remodelling (data not shown).
DISCUSSION
Exaggerated airway narrowing in response to bronchoconstricting stimuli is a defining feature of asthma. AHR is characterised by both an increase in sensitivity to such stimuli as well as an increase in the maximal airway narrowing that can be achieved [30]. Despite its importance, the mechanism(s) leading to AHR remain unclear. It has been suggested that changes in ASM phenotype could be responsible but nonmuscle properties of the airways have also been implicated [2]. A number of investigators have attempted to determine whether asthmatic ASM is mechanically different to nonasthmatic ASM [3–14], but the results thus far have been equivocal.
The present study provides evidence that there may be intrinsic differences in ASM behaviour in asthma, changes that could contribute to AHR. Although we found no differences in the stress produced by EFS, the velocity of shortening or the extent of shortening in the asthmatic muscle compared with the nonasthmatic muscle, there was a substantial difference in the relaxed muscle's response to a length perturbation. Although the active force generated by EFS was decreased following the length oscillation in both nonasthmatic and asthmatic tissues, the decrease was significantly less in the asthmatic tissue.
That an impaired response to length oscillation could contribute to AHR is based on evidence that the stretching of ASM as occurs in vivo during tidal breathing and deep inspirations (DIs) is sufficient to reduce ASM contractility, as demonstrated by in vitro experiments where ASM is subject to length oscillation [31, 32]. If this stretch-induced decrease in contractility were impaired, the response to any contractile stimulus could be enhanced (i.e. hyperresponsiveness).
However, it has recently been shown that oscillation amplitudes comparable to those produced by tidal breath (∼5 cmH2O) do not affect the response of airway segments to a contractile agonist, although with amplitudes >10 cmH2O the airways do respond with a transient dilation [33–36]. Moreover, the transient airway dilation observed in airway segments is short lived, compared with the longer lasting effect of DI-induced bronchodilation observed in healthy subjects [25, 37]. These data support the idea that the reduced airway response following deep inspiration is likely to be more complicated than a simple stretch of ASM.
We have previously shown that the reduction in force which follows a length oscillation is associated with a reduction in myosin thick filament density and we have suggested that it is this evanescence of myosin filaments that is responsible for the plasticity of the ASM length–tension relationship [38]. A similar change in smooth muscle function after deep inspiration in vivo could explain the bronchoprotective effect of DI. Our results support the possibility that the myosin thick filaments are altered in asthma, making them less prone to disruption following strain. Such an effect could prevent the decrease in force generation that accompanies repeated sighs and other forms of deep inspiration and could make the muscle more susceptible to exaggerated contraction.
The functional changes in smooth muscle that could contribute to AHR are an increase in force/stress generating capacity, increased shortening velocity, increased ability to shorten, the ability to adapt excessively at short lengths and/or the inability of length perturbations to allow plastic rearrangement of the contractile apparatus so as to decrease force production and shortening.
Our data suggests that asthmatic ASM does not produce more stress (force per unit muscle area) than nonasthmatic ASM. However, there was wide variation of stress produced in each group and a post hoc power analysis determined that ∼38 donors per group would be required to detect a 10% difference in mean stress between groups given this variation. While statistical significance may be possible with a larger sample size, it is not certain whether this level of difference in stress production would be physiologically relevant.
Our data suggest that a difference in shortening velocity is not an important contributor to AHR. A limitation of this conclusion is that we were technically unable to measure shortening velocity at very low loads. Using our apparatus we were not able to determine velocities at forces <2 mN, which was ∼5% Fmax. The study of larger strips of ASM may circumvent this limitation, i.e. Fmax would be large enough to avoid loads below 2 mN while working in the 5% Fmax range.
Using in situ length as reference length (Lref), both the asthmatics and the nonasthmatics had similar length–force relationships. Although the asthmatic ASM seemed to produce less force at the shorter and longer lengths than the nonasthmatics, these differences were not significant. However, following length adaptation, which was exhibited in both groups, the asthmatic ASM produced less force than the nonasthmatic ASM at the shortest length we examined (0.50 Lref). This observation suggests that asthmatic ASM is not more capable of length adaptation to short lengths than nonasthmatic ASM.
The different response to strain in the asthmatic ASM strips is the most intriguing aspect of this study (fig. 7). It is well known that asthmatics' airways respond differently to those of nonasthmatics to the stretch that accompanies deep inspiration [25, 39–42]. In vivo, the effects of deep inspiration can be assessed by taking big breaths before or after the administration of a bronchoconstricting stimulus. When applied after bronchoconstriction, DI produces a bronchodilating effect, while when taken before the administration of a constrictor, it causes a bronchoprotective effect, i.e. less constriction of the bronchi occurs when stimulated. Asthmatics may have a reduced bronchodilating effect of deep inspiration, especially during spontaneous attacks of asthma [43], but more consistently show a defective bronchoprotective effect such that prior deep inspiration fails to attenuate subsequent constriction [44–46].
Although it is tempting to speculate that the difference in attenuation in force following the length perturbation observed between asthmatic and nonasthmatic tissue is the basis for the reduced bronchoprotective response to DI in asthma, the picture is far from clear. While prior DI differentially modifies the methacholine-induced decline in forced expiratory volume in 1 s (FEV1) in asthmatics and nonasthmatics, it has no differential effect on the changes in FEV1/forced vital capacity ratio [47], partial expiratory flow [48] or airway resistance assessed using the forced oscillation technique [49]. It is argued by some that prior DI may not alter the initial airway narrowing produced by a constrictor, but instead makes the airway tree more responsive to a subsequent DI as is required to perform an FEV1 manoeuvre. Thus, the different response to stretch we observed is unlikely to be the sole mechanism to explain deficient bronchoprotection in asthma.
In the present study, the length oscillation was applied to the muscle before it was activated; this was to mimic the bronchoprotective effect of DI that has been shown to be more potent than the bronchodilating effect of DI in healthy human subjects [50]. Although, in isolated ASM preparations, length oscillation has been clearly shown to have a large effect on the muscle's subsequent ability to generate force [51], in airway segments, pressure oscillation does not lead to reduced airway narrowing on subsequent stimulation [52]. The cause for the discrepancy between the results with ASM strips and airway segments is not clear but it could be because mid-sized airways (used in the pressure oscillation experiments) are less distensible than the smaller airways. DI has been shown to reduce peripheral airway closure [47]. A recent study has shown that a reduction in airway distensibility is one of the reasons why severe asthmatics do not benefit from the bronchodilating effect of DI [53]. The reduced response of asthmatic ASM to length oscillation and the significantly higher passive tension at length greater than Lref observed in the present study could contribute to reduced airway distensibility.
In this study, we applied a length oscillation of 60% peak-to-peak, which means a 30% lengthening strain. Accounting for the compliance of the apparatus, this translates into an ∼25% lengthening of the smooth muscle preparation. This value of strain was selected because it resulted in significant force loss and decreased myosin thick filament density in previous studies [51]. However, this is more strain than is experienced by most of the ASM in the lung during a deep inspiration. If the airways dilate isotropically with the lung parenchyma, the circumferential strain is ∼20% with an inhalation from functional residual capacity (FRC) to total lung capacity (TLC), since FRC is ∼50% TLC. As most airways are less distensible than the lung, the strain that most of the smooth muscle experiences is <20%. We also applied a longer period of length oscillation than is used in vivo to detect bronchoprotection. Again this was chosen based on a previous protocol that was designed to maximise the effect of length oscillation on subsequent force development [51].
The difference in the response to DI in asthmatic subjects in vivo has been variously attributed to a failure of DI to translate into the same ASM strain in stiffened asthmatic airways or to an intrinsic difference in ASM mechanics. In vivo, deep inspiration applies a stress to the smooth muscle and the resultant strain is dependent on the stiffness of the airway wall as well as the elastic recoil of the lung (i.e. the same volume change produces less strain if lung recoil is reduced as it is in emphysema). Brown et al. [54] used computed tomography to estimate the strain (airway dilatation) produced by DI in normal and mild-to-moderate asthmatic subjects and found no difference. They suggested the different response to strain in asthma was at the basis of the defective response to DI. Our results support the contention that there is an intrinsic difference in smooth muscle behaviour in asthmatic tissue. We applied a length oscillation rather than a force oscillation. While this is nonphysiological, since strain is a dependent variable in vivo, it allows us to suggest that differences in the muscle response to strain, rather than attenuated strain due to stiff airways, is at the basis of the differential response.
Conversely, a similar strain experienced by an airway strip in vitro or by an airway in vivo could result in a different strain being applied to the contractile apparatus of the muscle. An airway strip or circumferentially stretched airway can be simplistically thought of as a contractile element in series with an elastic element. The relative strains experienced by the contractile and series elastic elements could be different for any overall strain.
Although we observed a lesser response to length oscillation in the asthmatic tissue, the active length tension curve before adaptation indicated that at high and low lengths force was lower in the asthmatic group (not consistent with AHR) and, importantly, there was no difference in adaptation. It seems paradoxical that the response to length oscillation differs but not the adaptive response. However, the difference in force recoveries (adaptation) after length oscillation and after a step change in length could be due to the fact that the recovery occurred at different lengths in the latter protocol. We observed that the asthmatic ASM tended to recover less at lengths shorter than Lref, and more at lengths greater than Lref. The reason for this difference is not clear. One can speculate that the structural disruption within the ASM associated with a 10-min length oscillation is different from that resulting from a step change in length, even though they both lead to some degree of force loss post-perturbation.
A possible explanation for the reduced response to stretch is that there could be persistent low-grade activation of the asthmatic muscle making it less responsive to stretch. However, such activation, if it did exist, was not manifested as an increased "passive" tension, as we did not observe an elevated resting tension in the asthmatic ASM (fig. 4). It could, however, manifest itself as an increased passive stiffness (which we did not measure) and that could also explain the higher passive tension (at 1.5 Lref) observed in the asthmatic group.
A strength of the present study is the well preserved tissue from asthmatic and nonasthmatic individuals. Some previous studies have been performed on airway tissue recovered at autopsy while others have examined specimens obtained at surgery. The study has limitations. 1) Little history was available on the subjects beyond that related to their terminal event. Because of this, we undertook a detailed morphological examination of the airways to quantify the extent of pathological changes that are typically found in asthma. The results showing that the fatal asthmatics, as well as those asthmatics who died of other causes, had features of airway wall remodelling provide an independent confirmation of the clinical phenotyping of the subjects. 2) The majority of the subjects had severe (fatal) asthma and thus the results may not apply to more mild disease. 3) The time from death to the time of study of the ASM was quite long, which could result in the loss of mediators present in life that could cause increased ASM contractibility. 4) Although the histological features of asthma were present in the intraparenchymal airways, the mechanics were performed only on the trachealis; the assumption that the properties of the ASM are uniform may not be valid. 5) The patient group is small and varied. Treatment before death for the asthmatic group ranged from none to prednisone. This variation in severity may have affected our ability to detect differences between the groups, but is unlikely to be responsible for the significant differences that we did find.
In conclusion, this study is the first to systematically examine the complete array of mechanical properties of ASM from asthmatic and nonasthmatic individuals. While the sample size was relatively small and the variability of some variables was wide, we found no substantial difference in the stress produced by the asthmatic muscle preparations; indeed at lengths longer and shorter than Lref, maximal stress tended to be somewhat less in asthmatic airway smooth muscle preparations. There was no difference in the velocity or extent of shortening.
Conversely, the results show, for the first time, that there is a difference in the ASM response to stretch in asthma. Following a 25% length oscillation there was less attenuation in ASM force in the asthmatic tissue. These results suggest that there is an intrinsic difference in the ASM's response to strain in asthma. The increased passive stiffness of the asthmatic preparations may, in part, contribute to this difference, which is supported by in vivo data showing less airway dilation induced by a DI in asthmatics [55]. Another possibility is that the myosin thick filaments in asthmatic ASM are less prone to dissolution and rearrangement.
Acknowledgments
The authors wish to acknowledge the technical assistance of D. Solomon, M. Elliott, D. English and A. Samra (all from the James Hogg Research Center, St Paul's Hospital, University of British Columbia, Vancouver, BC, Canada), as well as the statistical assistance of T. Lee (Center for Health Evaluation and Outcomes Sciences, Vancouver).
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This work was supported by operating grants from Canadian Institutes of Health Research (CIHR) (MOP-13271, MOP-4725). L.Y.M. Chin is supported by an Alexander Graham Bell Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC). Y. Bossé is supported by a fellowship from Fonds de la Recherche en Santé Québec (FRSQ) and a CIHR Strategic Training Initiative in Health Research-IMPACT fellowship. T.L. Hackett is supported by a Canadian Lung Association/CIHR/GSK, CIHR Strategic Training Initiative in Health Research-IMPACT and Michael Smith Foundation for Health Research Fellowships.
Statement of Interest
Statements of interest for T. L. Hackett and P.D. Paré can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received April 15, 2011.
- Accepted November 10, 2011.
- ©ERS 2012
REFERENCES










