





Abstract
We determined lung bioavailability of a fluticasone propionate (FP) pressurised metred-dose inhaler (Flovent® HFA; GlaxoSmithKline, Research Triangle Park, NC, USA) administered via AeroChamber Plus® (Monaghan Medical, Plattsburgh, NY, USA) with Facemask and Babyhaler® (GlaxoSmithKline) valved holding chambers (VHCs) using a population pharmacokinetic approach.
Children from 1 to <4 yrs of age with stable asthma but a clinical need for inhaled corticosteroid therapy were administered 88 μg FP hydrofluoroalkane (2×44 μg) twice daily delivered through the two devices in an open-label, randomised crossover manner for 8 days each. Patients were randomised to one of three sparse sampling schedules for blood collection throughout the 12-h dosing interval on the 8th day of each treatment for pharmacokinetic analysis. The area under the FP plasma concentration–time curve (AUC) was determined for each regimen.
17 children completed the study. The population mean AUC following FP with AeroChamber Plus® with Facemask was 97.45 pg·h·mL−1 (95% CI 85.49–113.32 pg·h·mL−1) and with Babyhaler® was 51.55 pg·h·mL−1 (95% CI 34.45–64.46 pg·h·mL−1). The relative bioavailability (Babyhaler®/AeroChamber Plus®) was 0.53 (95% CI 0.30–0.75).
Clinically significant differences in lung bioavailability were observed between the devices. VHCs are not interchangeable, as differences in drug delivery to the lung may occur. A population pharmacokinetic approach can be used to determine lung bioavailability of FP.
Valved holding chambers (VHCs) are essential for delivery of inhaled drugs from pressurised metered-dose inhalers (pMDIs) to the lungs in young children (<5 yrs of age) with asthma, and are frequently used in older children who have difficulty coordinating actuation and inhalation from a pMDI. However, delivery of a drug from a VHC to the lungs may be increased or decreased depending upon the specific VHC used (size, shape, plastic versus antistatic composition and dead-space volume), drug formulation (drug, propellant and excipient) and patient characteristics (tidal volume, calm versus crying and facemask seal) [1–5]. Ideally, the most relevant measure of drug delivery to the lungs is the clinical effect of the drug administered, but a direct relationship between the amount of drug delivered and response is nearly impossible to measure because response is influenced by other factors, including asthma severity, baseline pulmonary function, race/ethnicity, pharmacogenetic influences and duration of asthma [6]. In addition, for most inhaled drugs, there is no way to measure the activity of the drug that is due to direct delivery to the lungs from the activity of drug that is swallowed and absorbed from the gastrointestinal tract (oral bioavailability) without using activated charcoal to block gastrointestinal absorption or using scintigraphic imaging of labelled drug in the lungs. Studies involving activated charcoal or scintigraphic imaging are, at best, difficult to perform in young children, and such interventions are not ethical in this age group. Therefore, an alternative method to determine drug delivery to the lung is to measure blood levels after an inhaled dose. However, this method is suitable only for drugs that have negligible oral bioavailability to ensure that blood measurements reflect only drug that has been absorbed systemically from the lungs. Several published studies have documented differences in the delivery of inhaled corticosteroids (ICSs) to the lung with a metered-dose inhaler (MDI) attached to different VHCs by measuring blood levels [7, 8].
Fluticasone propionate (FP) was the most widely prescribed ICS in 2010 [9] and is often used with a VHC in the management of paediatric asthma. The oral bioavailability of FP is <1%, owing to nearly complete first-pass metabolism [10, 11]. Therefore, gastrointestinal and buccal absorption after inhaled delivery of FP do not contribute to systemic bioavailability [12]. Plasma concentrations following an inhaled dose of FP thus represent only drug that has been delivered to and absorbed systemically from the lungs (lung bioavailability). The area under the FP plasma concentration–time curve (AUC) and maximum plasma concentration are directly proportional to the inhaled dose [13]. Thus, FP is ideally suited to determining lung delivery from a VHC by measuring plasma concentrations after an inhaled dose.
We used a population pharmacokinetic approach in young children with asthma to compare lung bioavailability of FP (Flovent® HFA pMDI; GlaxoSmithKline, Research Triangle Park, NC, USA) delivered via two VHCs with facemasks: AeroChamber Plus® (Monaghan Medical, Plattsburgh, NY, USA) and Babyhaler® (GlaxoSmithKline).
METHODS
Study participants
Male or female children with physician-diagnosed asthma, aged 1 to <4 yrs at the start of randomisation, were recruited from two clinical sites (Nemours Children’s Clinic, Jacksonville, FL, USA and The University of Florida Paediatric Pulmonary Clinic, Gainesville, FL, USA). All children had a history of symptomatic asthma, and experienced at least two episodes of symptoms of asthma requiring medical attention and asthma pharmacotherapy within the 12 months preceding the screening visit. Children were eligible for participation if: they had required therapy with a maintenance asthma medication (including ICSs) and used the medication on a regular basis for the 4 weeks prior to the screening visit; or, according to their disease process and current treatment guidelines, should have been on ICS therapy (thus, demonstrating a clinical need for ICS therapy), and who planned to remain on this therapy. All corticosteroids (topical and inhaled) were discontinued at randomisation. Participants were allowed to remain on their prescribed nonsteroidal asthma medications during the study. Children were ineligible for participation if they: had an unresolved upper or lower respiratory tract infection within 2 weeks of the screening visit; had evidence of pneumonia within the preceding 3 months; had other significant pulmonary disease; or required oral corticosteroids within the 4 weeks prior to screening. Participants were not permitted to take any drug or food that might have inhibited or induced activity of cytochrome P450 3A4 within 2 weeks of dosing (e.g. erythromycin, phenobarbital and grapefruit juice). The participant’s parent, guardian or caregiver (referred to herein as “parent”) completed diary cards daily, and recorded date and time of dosing, adverse events, and concomitant medications. Institutional review board approval was obtained for each site. A signed and dated written informed consent/parental permission was obtained from each participant’s parent prior to the study.
Study design
This was a randomised, multiple-dose, open-label, two-way crossover study (GlaxoSmithKline protocol identifier FAS10002). Each participant had a screening visit conducted ≤30 days prior to randomisation. At the screening visit, medical history was obtained, a physical examination including vital signs was performed and blood was drawn for haematological analysis. Each child was randomised to receive a FP hydrofluoroalkane (HFA) pMDI attached to the AeroChamber Plus® with Facemask or Babyhaler® (identified as session 1 or 2). Children were administered two inhalations of FP, 44 μg per inhalation product actuated into the device 30 s apart twice daily. Doses were administered with a minimum of 11 h between the morning and evening doses for 8 days (176 μg total daily dose of FP). The first dose of each treatment was administered at the study site by the parent after instruction from the study coordinator. To ensure 100% adherence and steady state conditions, the final four doses of each session were observed by the study staff by having the parent and child return to the clinic or by the study staff going to the home of the participant to observe dosing. Blood for pharmacokinetic analysis was drawn on the 8th day of dosing. Following completion of day 8 study procedures, the participant was crossed over to the alternate regimen. After the final dose at the end of the study, the physical examination and blood draw for haematology analysis were repeated. There was no washout of prior inhaled corticosteroid dosing before the first dose of the first treatment or before crossover to the alternate regimen because the half-life of all ICSs used at the time of the study was sufficiently short after an inhaled dose that all drug would have been eliminated by the time of the pharmacokinetic sampling on day 8 [14].
Parents were trained on proper preparation of the MDI and VHC according to the product labelling instructions for use, and instructed that if the child became upset, began to cry, or fought the mask or holding chamber that dosing was to be delayed until the child was calm.
The parent recorded the date and time of dosing of study medication on the diary card. During treatment with the AeroChamber Plus® with Facemask, parents were instructed to have their child take six tidal breaths after each actuation. During treatment with the Babyhaler®, children took 5–10 tidal breaths after each actuation. A single study coordinator at each centre administered the doses on day 8 of sessions 1 and 2 in the clinical research facilities to reduce variability in administration technique between participants.
VHC devices
There are important design differences between the AeroChamber Plus® with Facemask and the Babyhaler®. The AeroChamber Plus® with Facemask is a 149-mL polycarbonate VHC that is 14.9 cm in length with a 4.5-cm fixed diameter and an integrated facemask (personal communication from Monaghan/Trudell Medical International, London, ON, Canada). The mask dead space ranges 24.0–46.0 mL depending upon the force applied to the mask on the child’s face [3]. There is a one-way valve between the VHC and the mask and an expiratory valve in the mask [15]; thus, there is no dead space between the valves [16]. The Babyhaler® is a 350-mL polycarbonate VHC that is 30 cm in length with a detachable facemask [1, 17]. The Babyhaler® has 40 mL of dead space between the low-resistance inlet and outlet valves, and 77-mL mask dead space for a total dead space of 117 mL [1, 15, 17]. The Babyhaler® weighs 190.7 g (mask and chamber) compared with AeroChamber Plus® with Facemask, which weighs 71.7 g (mask and chamber) [1].
At the Nemours Children’s Clinic, both AeroChamber Plus® with Facemask and Babyhaler® were prepared for first use according the manufacturers’ instructions, which for both devices, includes washing in soapy water, rinsing in clean water and air drying. At the University of Florida, the chambers were not washed. However, 28 doses were administered during each session, so the chambers were essentially primed at both sites prior to day 8 blood sampling.
Pharmacokinetic study design
Blood samples were taken for pharmacokinetic analysis on day 8 of sessions 1 and 2 according to one of three sampling schemes employing a standard population pharmacokinetic approach (table 1) [18]. Participants were randomly assigned to one of three sampling intervals, as follows. Group 1 (n=9): pre-dosing to 4-h post-dosing; group 2 (n=7): 2.5-h post-dosing to 8-h post-dosing; group 3 (n=2): 7.5-h post-dosing to 13-h post-dosing. The sampling intervals for groups 1, 2, and 3 reflected plasma concentrations due to absorption and distribution, distribution and elimination, and elimination and absorption following the next dose, respectively. The group assignment of a participant remained the same for both devices. Blood sampling was performed in the clinical research unit at the University of Florida and the Clinical Research Center at Wolfson Children’s Hospital (Jacksonville, FL). Both clinical research facilities are controlled environments with a stable daily temperature and humidity. The outdoor weather conditions were the same around the two centres where the participants lived.
Five 5-mL blood samples were drawn into a lithium heparin collection tube and stored on ice for ≤2 h after collection until they could be centrifuged at 4°C. Samples were drawn through an indwelling catheter (1-mL waste sample was discarded prior to study sample collection) or by needle stick. The use of topical anaesthetic (EMLA Cream; AstraZeneca LP, Wilmington, DE, USA) was permitted when drawing blood samples.
Plasma samples were frozen and stored at -20°C until they were analysed. Samples were analysed within 4 months of collection because stability data for plasma FP (stored at -20°C) were validated for only 4 months at the time of the study. Plasma FP was analysed by York Bioanalytical Solutions (York, UK) with a validated assay using solid-phase extraction in combination with liquid chromatography–tandem mass spectrometry, with an FP detection limit of 5 pg·mL−1 [19].
Pharmacokinetic analysis
The population pharmacokinetic model, using nonlinear mixed-effect modelling, was developed using the computer program NONMEM version V [18, 20, 21]. A one-compartment model with zero-order absorption and first-order elimination was developed, in which the lungs are believed to act as FP depot from which the drug is released into the systemic circulation. More complex models were assessed but did not appreciably improve fit.
Because a number of concentrations were not quantifiable, NONMEM was used to predict concentrations for unquantifiable values using a maximum likelihood method [22]. Additionally, pharmacokinetic parameters for individual participants were calculated using IPRED command in NONMEM. Because the sample size was small, the influence of covariates was not assessed [23].
Apparent clearance (CL/F) and relative bioavailability (F1) were obtained from the population pharmacokinetic model. The AUC was derived using the following equation:
A sample size of 14 participants was required to provide 90% power for the geometric means of the AUCs to be within 30% of each other with a confidence interval for the ratio contained within 0.70–1.43, based on a standard deviation for the difference in log-transformed values of 0.3134 and α=0.05 [7]. Additional participants, beyond the required 14, were randomised to the two sequences and assigned to the group 1 pharmacokinetic sampling scheme to maximise data when measurable levels were most likely.
RESULTS
Study participants
Of the 27 children that were screened, 20 were randomised and dosed. All randomised participants were included in the evaluation of the study population. A total of 17 children completed both sessions of the study and plasma FP data from these participants were included in the pharmacokinetic analysis. One participant completed both sessions but at the end of session 2 (day 8 with AeroChamber Plus® with Facemask), venous access was unsuccessful and, therefore, plasma samples could not be obtained for that session. Data from session 1 (Babyhaler® session) for this participant (group 2 sampling interval) was included in the population pharmacokinetic analysis. Two participants (each from session 1 and both during the Babyhaler® session) withdrew from the study prior to pharmacokinetic sampling due to other reasons not related to the study or due to an adverse event. The latter experienced a catheter-related complication (considered an adverse event) following administration of 88 μg FP HFA MDI b.i.d. with Babyhaler® that led to withdrawal from the study.
The mean age of the participants was 2.3 yrs (table 2). There were no serious adverse events reported. Of the seven participants who were screened but not randomised, three withdrew consent, three had abnormal laboratory results and one was lost to follow-up.
The mean±sd tidal volume of the 17 participants (tidal volume determined as 7 mL·kg−1 × ideal body weight [2]) was 100.3±17.0 mL (range 75.6–133.0 mL).
Pharmacokinetics
Plasma concentrations were measurable for ≤8 h following treatment with FP with the AeroChamber Plus® with Facemask and for ≤5 h with the Babyhaler® (fig. 1). The population mean AUC0–12 h was 51.55 pg·h·mL−1 (95% CI 34.45–64.46 pg·h·mL−1) with the Babyhaler® and 97.45 pg·h·mL−1 (95% CI 85.49–113.32 pg·h·mL−1) with the AeroChamber Plus® with Facemask. The relative bioavailability (Babyhaler®/AeroChamber Plus®) was 0.53 (95% CI 0.30–0.75).
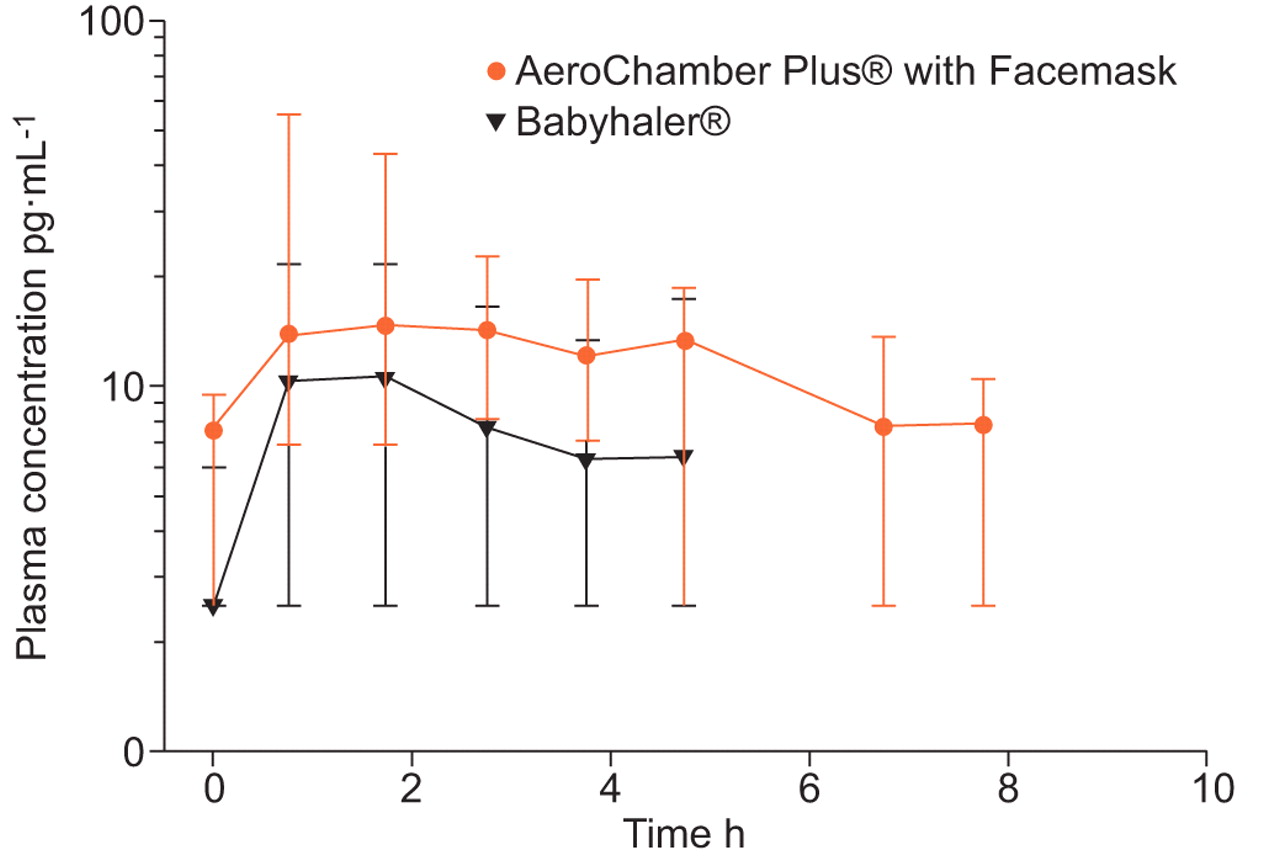
{kind=link}
Median plasma fluticasone propionate (FP) concentrations and 95% confidence intervals on a logarithmic scale after AeroChamber Plus® with Facemask (Monaghan Medical, Plattsburgh, NY, USA) and Babyhaler® (GlaxoSmithKline, Research Triangle Park, NC, USA). FP concentrations were measurable for a longer period of time after administration via AeroChamber Plus® with Facemask. A censored value of 2.5 pg·mL−1 was assigned when the concentration was below the limit of detection in order to calculate the median and 95% confidence interval.
DISCUSSION
The results of this study demonstrated that measurement of steady-state plasma concentrations following therapeutic doses of FP in preschool-aged children can reveal differences in lung bioavailability when doses are administered via different VHCs. This is the first study to directly compare lung bioavailability of ICSs delivered by two different VHCs in a crossover design in children aged 1 to <4 yrs using a population pharmacokinetic approach. Our findings confirm the results of previous studies in young and older children in which differences in lung bioavailability, expressed as peak concentration, were observed when FP was delivered via different VHCs [7, 8].
The AUCs observed in the present study (Babyhaler® 51.55pg·h·mL−1; AeroChamber Plus® with Facemask 97.45 pg·h·mL−1) were lower than that observed in two previous studies with similar patient populations. 88 μg FP twice daily or placebo was administered by AeroChamber Plus® with Facemask in one study (AUC 151 pg·h·mL−1) [24] and by Babyhaler® in the second study (AUC 141 pg·h·mL−1) [25]. It is unclear why the AUCs were lower in our study but differences in pharmacokinetic methods may provide one explanation. In the North and South American study with AeroChamber Plus® with Facemask [24], one or two samples were collected at defined intervals between 30-min and 9-h post-dose in each participant, and in the Babyhaler® study conducted in Europe and Australia [25], only one sample was collected from each participant at defined intervals between 15-min and 11-h post-dose. In our study, four post-dose samples were collected from each participant with each device. Thus, in our study, both devices were compared in the same population of participants and more frequent sampling was employed; therefore, there was greater control over variability. Also, in the Babyhaler® study [25], the lower limit of detection of FP plasma concentrations was 10 pg·mL−1, whereas in our study it was 5 pg·mL−1. Another possible explanation is that there may have been differences in FP plasma clearance as a result of differences in patients, ethnicity, diet and other factors. That is, clearance may have been higher in our study participants and, thus, AUC was lower. Another possible explanation for lower AUCs in our study is the duration of dosing: 8 days in our study and 12 weeks in the other two parallel studies [24, 25]. Whelan et al. [26] suggested that in adults, FP plasma concentrations may accumulate over a 6-week period, but it is unknown if this observation is reproducible and whether it applies to young children. However, while it is possible that the shorter duration in our study explains the lower AUCs, this would not have any effect on relative bioavailability, because we used a crossover design of short duration in which the order of each device (AeroChamber® first or Babyhaler® first) occurred equally in the study.
The lower AUC from the Babyhaler® versus the AeroChamber Plus® with Facemask is probably due to differences in device characteristics. The Babyhaler® is larger in volume with greater dead space and requires more tidal breaths (approximately four to six breaths) to empty the chamber than the AeroChamber Plus® (approximately two breaths) for children with a tidal volume of 180–195 mL, according to in vitro studies [15, 27]. The tidal volumes calculated for the children in this study ranged 76–133 mL [2], which are less than the tidal volume used in the in vitro simulation studies [15, 27]. This suggests that even more breaths would be required to empty the Babyhaler® compared with the AeroChamber Plus® with Facemask. It is possible that the increased time required for the additional breaths with the Babyhaler® could result in greater drug fallout within the chamber, which would reduce the drug available for inhalation [16].
For practical and ethical reasons, we did not measure the actual dose delivered to the patient. Several in vitro studies using a lung model appropriate for children 1–4 yrs of age have demonstrated that the amount of drug available for lung delivery to the patient is similar between the Babyhaler® and the AeroChamber® or Aerochamber Plus® [15, 16, 27], and is approximately 17–24% of the delivered dose. However, in vitro models may not accurately reflect differences in lung delivery in vivo, as illustrated by the results of our study.
At the Nemours Children’s Clinic, VHCs were prepared for use according to the manufacturers’ instructions, which include washing in soapy water, rinsing in clean water and air drying. The soapy water was not allowed to air dry in the VHC, which is a known method to reduce electrostatic charge [28, 29]. At the University of Florida, the chambers were used out of the package without conditioning. However, at both sites, each VHC had 28 doses discharged into the device before day 8, and previous studies have shown increased lung delivery of salbutamol and budesonide by priming the VHC with 10–20 actuations to reduce the electrostatic charge [30, 31]. In an in vitro model that incorporated tidal volume, chamber volume, dead-space volume, effects of valve insufficiency or other leaks from the VHC, aerosol fallout within the chamber and aerosol loss from immediate impact on the VHC wall, priming was found to have the greatest effect on the dose available for inhalation [15]. While the VHCs were, in effect, primed prior to day 8 with twice daily dosing, it is possible that due to the larger size of the Babyhaler®, >28 actuations into the Babyhaler® would be required to reduce the electrostatic charge; thus, incomplete priming may have contributed to the lower AUC from the Babyhaler® [16].
Facemask seal has been shown to be an important determinant of the amount of drug inhaled [4, 32–34]. However, no parent noted any difficulties with the Babyhaler® that might have affected drug delivery. In addition, the last four doses were administered by the parent in the presence the study coordinator, and the day-8 dose was administered by the same study coordinator for all patients at each site to reduce intra- and interpatient variability. Dose counters were not available on the FP canisters at the time of the study and other measures of adherence (patient report and weighing canisters) are not accurate for assessing adherence [35, 36].
Blood samples were collected using a population pharmacokinetic approach. Several population pharmacokinetic blood sampling designs could have been used, but we chose to use a sparse sampling technique with mixed-effect modelling in order to minimise the number of blood samples required from each participant. This methodology is particularly attractive for paediatric pharmacokinetic studies [24, 25, 37, 38]. With this design, children are allocated to one of several blood sampling schemes, such that the entire dosing interval is sampled but no one child is subjected to blood sampling at each time-point. This method uses the aggregate population for the analysis rather than data from the individual. Mixed-effect modelling describes the data using both fixed and random effects to describe the intersubject variability [18, 23]. In the present study, the influence of covariates was not assessed due to the relatively small number of participants. A one-compartment model with zero-order absorption and first-order elimination was found to fit these data best.
Despite a near doubling of the AUCs with the AeroChamber Plus® versus the Babyhaler®, adrenal suppression at a FP dose of 176 μg·day−1 (which is considered a low dose) [5, 39] is unlikely, except in a minority of patients [24, 25, 40–45]. It is possible that as airway obstruction improves, there will be greater lung delivery [46, 47] and potentially greater systemic effects, as changes in serum cortisol are dose-dependent [48, 49]. The maximal effect of FP on pulmonary function is reached in ∼3 weeks [50]; thus, effects on serum cortisol would be seen early in treatment. Importantly, noted decreases in serum cortisol or urinary cortisol excretion do not appear to translate into observable clinical adverse effects (reduced growth rate or ocular effects) with use at this dose (or an equivalent dose of other ICSs) for ≥1 yr [40, 41]. However, individual sensitivity to these effects has been observed [25, 44, 45]. In addition, a pharmacokinetic/pharmacodynamic model developed to define the relationship between systemic exposures (in cortisol equivalents) and changes in growth velocity, determined that no effect on growth velocity would be predicted with either VHC at the dose of FP used in this study [38].
Our study is limited by the relatively small sample size, which precluded evaluating the effect of covariates on AUC. The covariate most likely to have affected results would have been airway obstruction. However, spirometry cannot be performed in children this young, and oscillometry for measurement of airway resistance and reactance was not available at our sites; only a few centres in the USA have this procedure available. Systemic bioavailability of inhaled FP is greater in healthy adults (without airflow obstruction) compared with patients with asthma [46, 47]. However, we would not expect this to be a factor to explain the differences we observed between the Babyhaler® and the AeroChamber Plus® with Facemask. This was a crossover study of short duration, so changes in airway obstruction between treatment assignments that could have affected drug delivery to the lungs would have been unlikely.
Our results have several important implications for the dosing of ICSs in young children, as well as future research studies. It is clear from our study and other studies using different devices that VHCs are not interchangeable. This lung bioavailability study shows that depending upon the VHC chosen, a nearly two-fold difference in the ICS dose delivered to the lungs could occur. A two-fold difference in delivery has been suggested to be clinically relevant [51]. While the dose–response relationship of ICSs in adults is relatively flat and maximum response is achieved in most patients with low doses [48], dose–response relationships have not been well characterised in young children. Thus, if a child is clinically stabilised on a low ICS dose and switches to a VHC with two-fold lower lung delivery, asthma control could worsen. Conversely, a child maintained on a medium or high ICS dose (e.g. ≥352 μg·day−1, FP-equivalent, in 0–4-yr-old children) could experience adverse systemic effects if switched to a VHC with two-fold greater delivery. Parents, clinicians and pharmacists should be educated not to interchange VHCs once a child is stable on a particular ICS dose and VHC combination. Moreover, the initial prescription for a VHC should include language (e.g. “Do not substitute” or “Medically necessary”) to prevent the pharmacist from substituting a different VHC. We suggest that each VHC should be evaluated for relative lung bioavailability prior to their routine use with a particular ICS.
Acknowledgments
Our thanks go to D. Collins, S. Srebro and L. Sutton (GlaxoSmithKline, Research Triangle Park, NC, USA) for their review of this manuscript.
Footnotes
Support Statement
This study was supported by a grant from GlaxoSmithKline (Research Triangle Park, NC, USA) and National Institutes of Health General Clinical Research Center grant M01 RR00082.
Statement of Interest
Statements of interest for R. Mehta, R.L. Kunka and L. Hendeles, and for the study itself can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received December 1, 2010.
- Accepted June 15, 2011.
- ©ERS 2012
REFERENCES










