





Abstract
Haematopoietic c-kit+ progenitor cells may contribute to pulmonary vascular remodelling and pulmonary hypertension (PH). Stromal derived factor-1 (SDF-1/CXCL12) and its receptors CXCR4 and CXCR7 have been shown to be critical for homing and mobilisation of haematopoietic c-kit+ progenitor cells in the perivascular niche.
We administered AMD3100, a CXCR4 antagonist, and CCX771, a CXCR7 antagonist, to chronic hypoxia exposed mice in order to study the role of c-kit+ progenitor cells in PH. CXCL12, CXCR4 and CXCR7 protein expression, haemodynamic parameters, right ventricular mass, extent of vascular remodelling and perivascular progenitor cell accumulation were studied.
Chronic hypoxia-exposed mice showed increased total lung tissue expression of CXCR4, CXCR7 and CXCL12 after development of PH. This was associated with significantly increased right ventricular systolic pressure and evidence of right ventricular hypertrophy, vascular remodelling and perivascular c-kit+/sca-1+ progenitor cell accumulation. CCX771 administration did not abrogate these effects. In contrast, administration of AMD3100, whether alone or combined with CCX771, prevented vascular remodelling, PH and perivascular accumulation of c-kit+/sca-1+ progenitor cells, with a synergistic effect of these agents.
This study offers important pathophysiological insights into the role of haematopoietic c-kit+ progenitors in hypoxia-induced vascular remodelling and may have therapeutic implications for PH.
Many studies in recent years have demonstrated that non-resident progenitor cells may contribute to chronic hypoxia-induced pulmonary hypertension (PH) [1]. There is accumulating evidence for the existence of bone marrow (BM)-derived circulating endothelial progenitor cells [2], circulating smooth muscle progenitor cells [3] and circulating fibrocytes [4], which may contribute to the process of vascular remodelling. Currently, there is no definitive consensus on the single best marker for BM-derived vascular progenitor cells. However, a generally accepted marker for BM-derived haematopoietic progenitor cells is c-kit/CD117, a transmembrane tyrosine kinase receptor for stem cell factor [5]. Previous studies have reported that c-kit+ cells are mobilised from the BM and tracked to atherosclerotic lesions [6] and sites of myocardial infarction [7]. Moreover, Toshner et al. [8] have shown the presence of cells expressing progenitor markers particularly within the plexiform lesions associated with severe pulmonary arterial hypertension in humans. This was accompanied by the expression of progenitor homing signals in these lesions.
The CXC chemokine stromal derived factor-1 (SDF-1/CXCL12) and its receptor CXCR4 play a major role in regulating cancer cell metastasis [9], lymphocyte trafficking [10] and mobilisation of haematopoietic progenitor cells to ischaemic tissue [11]. Recent evidence indicates that there is another seven transmembrane CXCL12 receptor encoded by RDC1/CXCR7 [12, 13]. CXCR7 may complex with CXCR4, forming a heterodimer that potentiates CXCL12 signalling [14]. Surface expression of CXCR7 was reported on tumour cells, activated endothelial cells, fetal liver cells and other cell types [15]. It has been suggested that CXCR7 regulates several major biological processes including cell survival, cell adhesion and tumour development in animal models [15]. Expression of CXCR4, CXCR7 and CXCL12 is upregulated in response to tissue hypoxia and contributes to the recruitment of circulating progenitor cells [16–18].
Accordingly, the aim of the our study was to determine if administration of AMD3100, a CXCR4 antagonist, CCX771, a CXCR7 antagonist, or both would attenuate hypoxia-induced PH by reducing pulmonary c-kit+ haematopoietic progenitor cell accumulation.
MATERIALS AND METHODS
Study design
Preventative strategy
44 6-week old male C57BL/6J mice (Janvier, Le Genest-St-Isle, France) were exposed to normobaric hypoxia (10% inspiratory oxygen fraction (FI,O2)) or normoxia (20.9% FI,O2) for 3 weeks and then randomly assigned to four groups to receive daily: 1) intraperitoneal injections of saline; 2) intraperitoneal injections of 10 mg·kg−1 AMD3100 (Sigma-Aldrich, Saint-Quentin-Fallavier, France); 3) subcutaneous injections of 10 mg·kg−1 CCX771 (Chemocentryx, Mountain View, CA, USA); and 4) combined treatments with AMD3100 and CCX771, both at 10 mg·kg−1 from day 1 to day 21.
Therapeutic strategy
28 6-week-old male C57BL/6J mice were exposed to normobaric hypoxia (10% FI,O2) or normoxia (20.9% FI,O2) for 5 weeks. After 3 weeks of this exposure, the mice were randomly assigned to receive daily: 1) intraperitoneal injections of saline; 2) intraperitoneal injections of 10 mg·kg−1 AMD3100; 3) subcutaneous injections of 10 mg·kg−1 CCX771; and 4) combined treatments with AMD3100 and CCX771, both at 10 mg·kg−1 from day 21 to day 35.
Animal experiments were approved by the administrative panel on animal care at Centre de Chirurgie Expérimentale Marie Lannelongue (Le Plessis-Robinson, France).
Haemodynamic measurements and tissue collection
Mice were anesthetised with intraperitoneal injection of ketamine (60 mg·kg−1) and xylazine (8 mg·kg−1). Blood samples were collected from the retro-orbital plexus. Right ventricular systolic pressure (RVSP) was recorded after insertion of a 24-gauge needle connected to a pressure transducer into the right ventricle via the right jugular vein. Mice were euthanised by exsanguination. Lungs were inflated by intratracheal infusion of 50% OCT in PBS, snap frozen and stored at -80°C. Right ventricular hypertrophy was assessed by calculating the weight ratio of the right ventricle (RV) to the left ventricle (LV) plus septum (S) (RV/(LV+S)).
Immunofluorescence
Staining was performed on 5 μm acetone-fixed frozen sections. The anti-mouse primary antibodies used were: fluorescein isothiocyanate-conjugated anti-α-smooth muscle actin (Sigma-Aldrich), biotinylated anti-CD184/CXCR4 (BD Biosciences, Le Pont-de-Claix, France), anti-RDC1/CXCR7 (Novus Biologicals, Littleton, CO, USA), anti-SDF-1α/CXCL12α (eBioscience, San Diego, CA, USA), biotinylated anti-c-kit (Abcam, Paris, France), anti-Sca-1 (R&D Systems, Lille, France), anti-FcϵRIα (immunoglobulin E high affinity receptor; eBioscience), and anti-mast cells tryptase (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Isotype-matched irrelevant antibodies were used as negative controls. AlexaFluor488-, AlexaFluor594- or AlexaFluor647-conjugated species-specific secondary antibodies or streptavidin were used to reveal the staining (Invitrogen, Cergy-Pontoise, France). Slides were observed under a routine fluorescence and confocal laser scanning microscope (Olympus FV-1000; Olympus, Rungis, France) with appropriate emission filters.
Microscopic morphometry
Medial thickness defined as the ratio of the medial wall thickness to the external diameter of pulmonary arterioles with a diameter <30 μm, 30–50 μm, 50–75 μm and 75–100 μm was measured on 30 arterioles per α-actin-stained slide at random. The mean number of c-kit+ cells per pulmonary artery (cross-sectional diameter of 30–100 μm) was calculated by counting c-kit expressing cells on 30 arterioles per mice (n = 3 per group).
Western blot
Western blot was performed on normoxic and hypoxic lungs (n = 3–5), as previously described [19]. Membranes were stained with anti-CXCR4 (Capralogics, Hardwick, VT, USA), anti-CXCR7 (Novus Biologicals) or anti-CXCL12 (eBioscience) antibodies then incubated with horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualised by Lumi-LightPLUS Western Blotting Substrate (Roche, Neuilly-sur-Seine, France) using New Hyperfilm ECL (GE Healthcare, Orsay, France). Membranes were stripped in the Restore Western Blot Stripping Buffer (Thermo Fisher, Brebières, France) and reused for detecting β-actin bands with a goat-polyclonal antibody (Abcam). Protein band intensity was determined using ImageJ (National Institutes for Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/).
Statistical analysis
Quantitative variables were presented as mean±sem. Between groups, multiple group comparisons were performed with ANOVA and the Fisher projected least significant difference method as a post hoc analysis. A p-value <0.05 was considered to reflect statistical significance.
RESULTS
CXCL12, CXCR4 and CXCR7 expression and perivascular haematopoietic progenitor cell accumulation in lungs after 3 weeks of hypoxia
There was a significantly increased expression of CXCR4, CXCR7 and CXCL12 in the total lung tissues of hypoxic mice (1.08±0.19, 0.55±0.02 and 1.52±0.25, respectively) compared with normoxic mice (0.37±0.07, 0.23±0.02 and 0.45±0.03, all p<0.05) (fig. 1a–d). This was associated with a strong immunostaining for CXCL12 in hypoxic pulmonary arterial endothelial and smooth muscle cells and for CXCR4 and CXCR7 in pulmonary arterial endothelial cells compared with normoxia. Moreover, chronic normobaric hypoxia was associated with a perivascular accumulation of CXCR4+ cells and c-kit+ cells around remodelled pulmonary arteries (fig. 1e–l). We examined the origin of these cells using haematopoietic and mast cell markers. Double immunostaining for c-kit, in combination with Sca-1 and CXCR4, respectively, confirmed the haematopoietic origin of these cells. Moreover, most accumulated cells were negative for FcϵRIα and mast cell tryptase. Double immunofluorescence for c-kit and α-smooth muscle actin (α-SMA) showed that some c-kit+ cells observed around pulmonary arterial lesions may co-express α-SMA, indicating a likely differentiation of these cells into myofibroblasts (fig. 2).

CXCL12, CXCR4, and CXCR7 expression and perivascular haematopoietic progenitor cell accumulation in lungs after 3 weeks of hypoxia. a–d) Overexpression of CXCL12 and its two receptors CXCR4 and CXCR7 in total lung protein extracts from hypoxic (H) mice compared with normoxic (N) mice measured by western blotting. *: p<0.05; ***: p<0.001. Expression is normalised towards β-actin. n = 3 in the control group versus n = 5 in the hypoxic group. e–l) Immunohistochemical analysis of vascular localisation of e, f) CXCL12 and their receptors g, h) CXCR4 and i, j) CXCR7, and k, l) accumulation of perivascular c-kit+ and CXCR4+ cells. Normoxia (e, g, i, k) and hypoxia (f ,h, j, l) mouse lung. Scale bars = 50 μm.

Characterisation of haematopoietic c-kit+ progenitor cells in pulmonary vasculature during hypoxia. Hypoxic pulmonary hypertension mice have shown a lung accumulation of c-kit+ cells. Most c-kit+ cells co-expressed the progenitor haematopoietic marker a) CXCR4 and b) Sca-1. Double immunofluorescent staining of c-kit for mast cell c) tryptase and d) FcϵRIα has shown most of c-kit+ cells to be negative for both of these markers. e) Some cells were positive for dual staining with c-kit and α-SMA, indicating a possible differentiation of these cells into myofibroblasts (arrowhead). Scale bars = 50 μm.
Effects of AMD3100 and CCX771 on hypoxia-induced PH
Haematocrit
We verified that haematocrit values were significantly increased in hypoxic mice on day 21 as well as day 35 compared with normoxic control mice (61.7±1.2 versus 46.0±1.4, p<0.0001). AMD3100 and CCX771 treatment had no effect on the haematocrit values observed in hypoxic animals (data not shown).
Haemodynamics and right ventricular hypertrophy
Preventative strategy
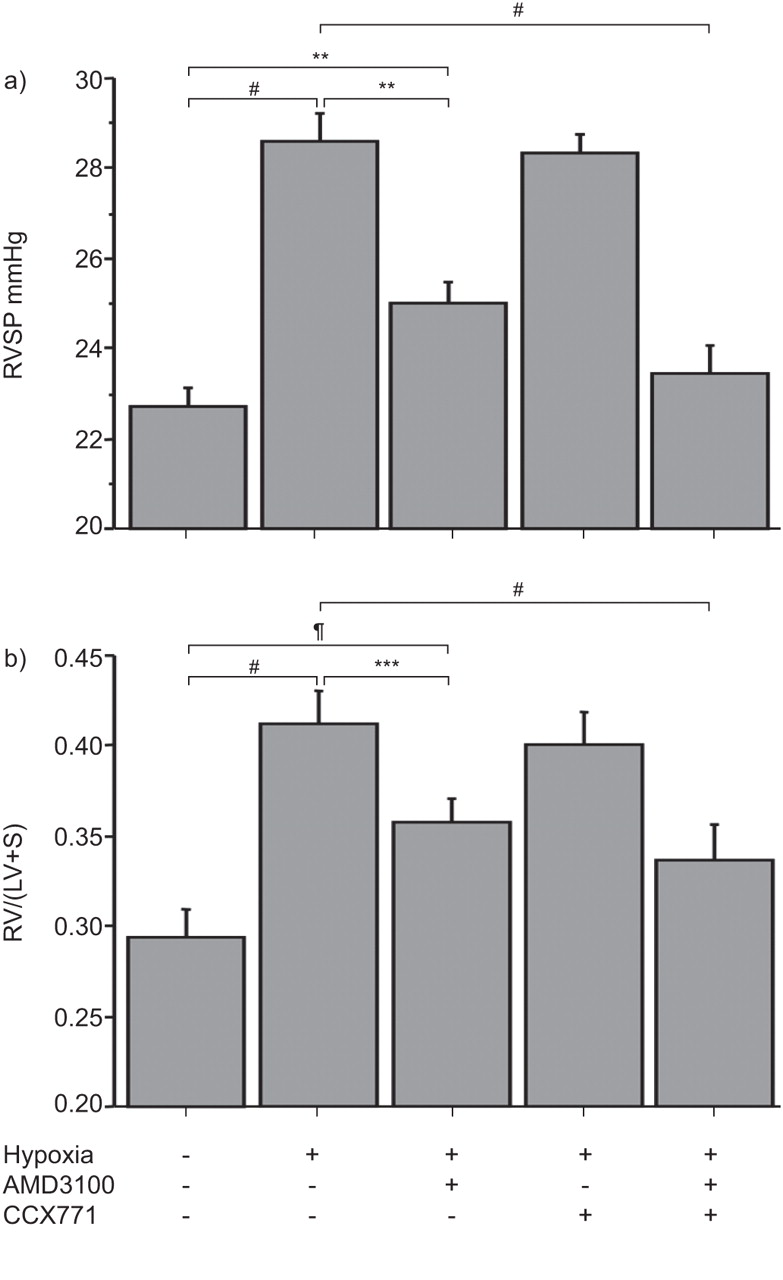
We then sought to elucidate whether inhibition of CXCR4/CXCR7 would prevent PH. The RVSP of hypoxic mice was significantly increased compared with normoxic mice (28.6±0.6 mmHg versus 22.7±0.5 mmHg, p<0.001). The RVSP of AMD3100-treated mice (25.0±0.5 mmHg) was substantially lower than the RVSP of the hypoxic mice (p<0.001) but still significantly higher than the RVSP of normoxic mice (p = 0.02), indicating a significant but partial effect of AMD3100. While CCX771 alone had no effect on hypoxia-induced PH (28.3±0.4 mmHg), a treatment combining AMD3100 and CCX771 induced a RSVP significantly lower than RVSP of hypoxic mice (23.4±0.6 mmHg, p<0.0001), indicating a synergistic effect of AMD3100 and CCX771. Accordingly, the RSVP in combined treatment with AMD3100 and CCX771 was not significantly different from RVSP of normoxic mice (p = 0.46) (fig. 3a).

Impact of AMD3100, CCX771 and their combination on haemodynamic and right ventricular hypertrophy in hypoxia-induced pulmonary hypertension. a) Right ventricular systolic pressure (RVSP) in the different treatment groups. b) Ratio of the right ventricle (RV) to the left ventricle (LV) plus septum (S) weight (RV/LV+S). #: p<0.0001; ¶: p<0.02; **: p<0.01; ***: p<0.001.
Hypoxia-induced PH was associated with a significant hypertrophy of the right ventricle, defined by the RV/(LV+S) ratio, compared with normoxic controls (0.41±0.02 versus 0.29±0.01, p<0.0001). In accordance with the beneficial effect of AMD3100 on RVSP in hypoxic mice, the RV/(LV+S) ratio was also significantly decreased in hypoxia-exposed AMD3100-treated mice (0.35±0.01). CCX771 alone had no effect on hypoxia-induced right ventricular hypertrophy (0.41±0.02) while combined treatment with AMD3100 and CCX771 was not statistically significant compared with normoxic mice (0.33±0.02, p = 0.14) (fig. 3b).
Therapeutic strategy
Exposure of adult mice to 5 weeks hypoxia also resulted in a marked increase in RVSP (28.8±0.5 mmHg versus 22.7±0.4 mmHg, p<0.0001) and in right ventricular hypertrophy (0.39±0.03 versus 0.29±0.04, p<0.0001) compared with normoxia. In contrast, administration of AMD3100, CCX771 or both for 2 weeks in mice with established PH had no significant effect on RVSP and RV/(LV+S) ratio (data not shown).
Effect of CXCR4 and CXCR7 antagonism on hypoxia-induced pulmonary vascular remodelling
We then studied the effects of CXCR4 and CXCR7 antagonism on pulmonary vascular remodelling in mice. Medial thickness of pulmonary arterioles <30 μm, 30–50 μm, 50–75 μm and 75–100 μm in hypoxic mice was significantly increased compared with normoxic mice. Medial thickness of pulmonary arterioles <30 μm, 30–50 μm, 50–75 μm and 75–100 μm in AMD3100-treated hypoxic mice was significantly decreased compared with the hypoxic control mice, but remained increased compared with the normoxic animals. CCX771 treatment alone had no significant effect on hypoxia-induced pulmonary arterial thickening. However, the medial thickness in hypoxic mice receiving a treatment combining AMD3100 and CCX771 was significantly lower compared with hypoxic control mice and hypoxic mice treated with AMD3100 or CCX771 alone. No difference was observed between the medial thickness in hypoxic mice treated with the combined AMD3100 and CCX771 treatment and medial thickness of normoxic mice (fig. 4).
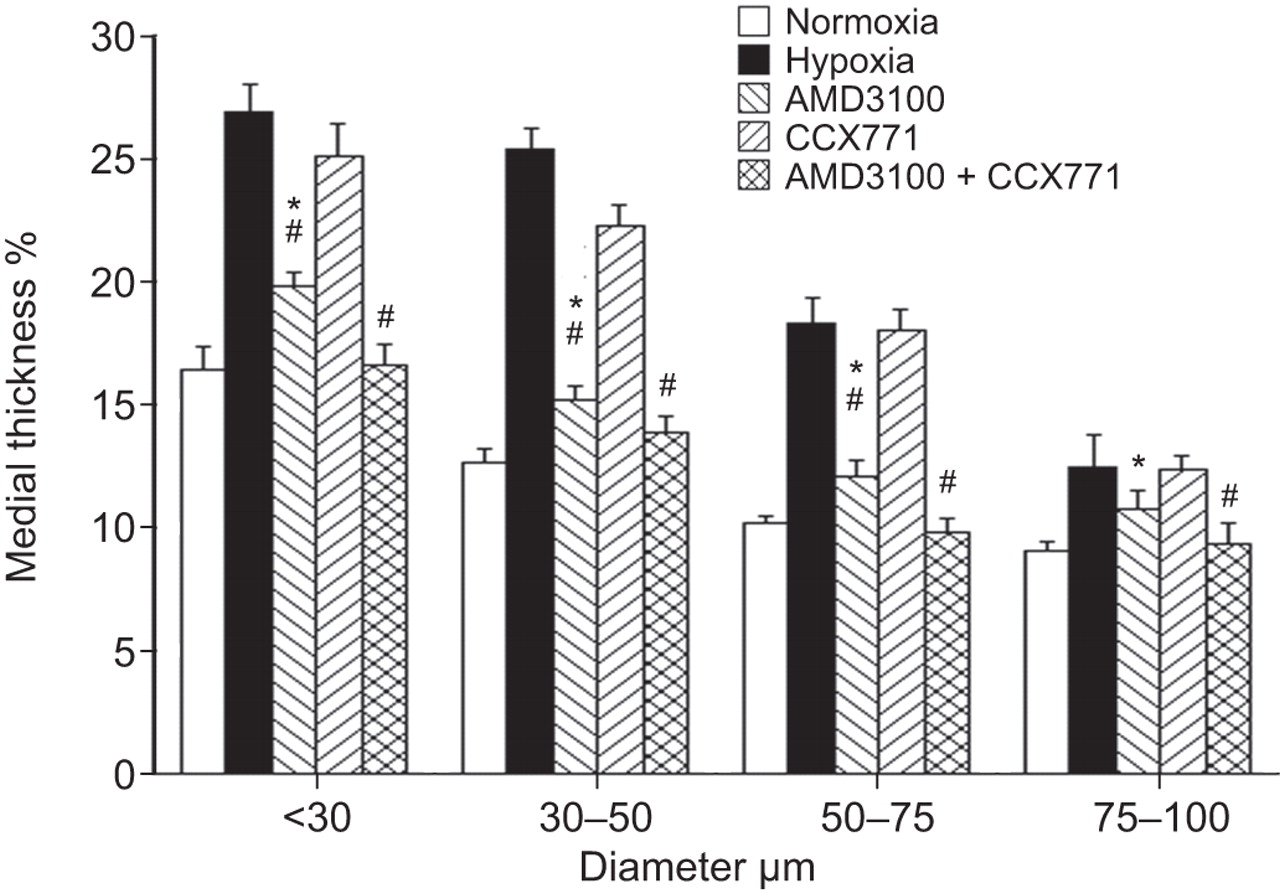
Effects of AMD3100, CCX771 and their combination on the medial wall thickness. 30 pulmonary arteries per mice (n = 3) were sized and separated into four groups: <30 μm, 30–50 μm, 50–75 μm and 75–100 μm. Each bar represents the mean ratio of the medial wall thickness to the external diameter of pulmonary arterioles, measured on ≥30 arteries from three slides. *: p<0.05 versus control; #: p<0.05 versus hypoxia at day 21.
Effect of CXCR4 and CXCR7 antagonism on hypoxia-induced perivascular c-kit+ progenitor cell accumulation
Hypoxia-induced pulmonary vascular remodelling was associated with a significant accumulation of c-kit+/FcϵRIα-/tryptase- progenitor cells distinct from the few c-kit+/FcϵRIα+/tryptase+ mast cells in the adventitia of remodelled pulmonary arteries compared with normoxia (3.8±0.1 versus 0.6±0.1 cells·artery−1, p<0.0001).
Given that CXCL12 is an integral component of progenitor cells mobilisation during hypoxia and that c-kit+ cells were increased in the lungs of adult mice with hypoxia-induced PH, we next evaluated if perivascular c-kit+ cell numbers could be decreased by inhibition of the CXCR4/CXCR7 pathway. AMD3100 therapy significantly decreased the number of c-kit+ cells compared with hypoxic mice (1.8±0.1 cells·artery−1, p<0.0001), although it remained significantly higher than in normoxic mice (p<0.0001). Combined AMD3100 and CCX771 treatment (1.0±0.1 cells·artery−1) was the most efficient in reducing hypoxia-induced c-kit+ progenitor cell accumulation compared with AMD3100 alone (p<0.0001) and normoxic mice (p = 0.016) (fig. 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of the c-kit+ cell infiltration in the vicinity of vascular lesions. Effect of CXCR4 and CXCR7 antagonism on hypoxia-induced perivascular accumulation of haematopoietic c-kit+ progenitor cells. #: p<0.0001.
DISCUSSION
We analysed the effects of the antagonism of two major chemokine receptors involved in the recruitment of haematopoietic progenitor cells in the setting of hypoxia-induced PH. We demonstrated that AMD3100, an antagonist of CXCR4, and its combination with CCX771, an antagonist of CXCR7, prevent, at least in part, pulmonary vascular remodelling, PH and right ventricular hypertrophy induced by chronic hypoxia. These effects were associated with a significantly decreased number of perivascular c-kit+ haematopoietic progenitor cells associated with hypoxia-induced PH.
Hypoxia is the main stimulus for the induction of PH in people who live at high altitude or PH complicating chronic respiratory disorders, such as chronic obstructive pulmonary disease and interstitial lung diseases. While acute hypoxia causes selective pulmonary arteriolar vasoconstriction, chronic exposure to hypoxia results in morphological and functional changes in the pulmonary vascular bed [20]. Hypoxia is a potent activator of the CXCR4/CXCL12 pathway. Of note, CXCL12 acting through CXCR4 is the main chemokine that modulates trafficking of haematopoietic stem and progenitor cells. Recently, it has been shown that CXCL12 plays a crucial role to mobilise and recruit BM-derived CXCR4+ proangiogenic cells to the hypoxic tissue. CXCL12 can act through CXCR4 on vascular endothelium [21], upregulating and activating adhesion molecules to promote stable interactions with circulating cells [22]. Moreover, recruited CXCR4+ cells may produce angiogenic cytokines that promote the proliferation and differentiation of resident vascular cells [11]. In our present study we extend, in adult hypoxic animals, recent data obtained in neonatal animals demonstrating hypoxia-induced perivascular CXCL12 and CXCR4 overexpression that can result in pulmonary artery adventitial c-kit+ haematopoietic progenitor cell accumulation [23, 24]. Young et al. [24] showed that the inhibition of the CXCL12/CXCR4 axis in the setting of lung development both inhibited and reversed pulmonary vascular remodelling in neonatal PH mice and this was associated with decreased stem cell lung expression. However, this study lacked precise identification and counts of progenitor cells. The count of c-kit+ cells was performed on whole images and not in the vicinity of remodelled vessels. In addition, no marker was used to exclude mast cells that also express c-kit. The c-kit+ cells localised in the parenchyma are likely to be mast cells co-expressing FcϵRIα or tryptase. We unequivocally demonstrated that inhibition of the CXCL12/CXCR4 axis decreased pulmonary vascular remodelling as well as the number of recruited pulmonary c-kit+/FcϵRIα- and c-kit+/tryptase- cells in an experimental model of PH, by directly counting these cells in the vicinity of remodelled small arteries that were categorised by size.
We also extended the study to the role of RDC1/CXCR7, another recently identified receptor of CXCL12 [12, 13]. Indeed, several reports indicate that CXCR7 plays a role in the response to hypoxia [25, 26]. CXCR7 may complex with CXCR4, forming a heterodimer that potentiates CXCL12 signalling [14]. It has been demonstrated that CXCR7 regulates several important biological processes including cell survival, cell adhesion and tumour development in animal models [15]. However, the mechanisms responsible for CXCR7 signal transduction are not completely understood. To date, the CXCL12/CXCR7 interaction has not been shown to activate G-protein-mediated signal transduction and it has been speculated that CXCR7 may clear secreted CXCL12 from the extracellular environment and thereby help maintain a defined CXCL12 gradient [27]. While AMD3100 had only a partial effect and CCX771 had no effect on hypoxia-induced c-kit+ progenitor cell accumulation and PH, we showed that combined CXCR4 and CXCR7 antagonism may have synergistic effects and prevent hypoxia-induced haematopoietic progenitor cell accumulation in the perivascular area and PH. These results may be explained by several recently described features of these pathways. Kalatskaya et al. [28] showed that AMD3100, mainly known as a competitive ligand for CXCR4, is also an allosteric ligand for CXCR7 at concentrations that are generally used by investigators in in vitro studies. Accordingly, in contrast to its negative effect on the CXCR4/CXCL12 interaction, AMD3100 may positively modulate CXCL12 effects and binding with CXCR7. Such a mechanism could participate in the synergy we observed by combined antagonism of the two receptors. In addition, AMD3100 recruits β-arrestin to CXCR7 but blocks β-arrestin recruitment to CXCR4. This might explain the two-fold decrease in haematopoietic c-kit+ cell accumulation and the partial improvement of vascular remodelling in the setting of AMD3100 monotherapy. Moreover, it could explain why these parameters were completely reversed with combined AMD3100 and CCX771 therapy. Thus, our study shows that blocking the CXCL12/CXCR4 axis is not sufficient to fully prevent hypoxic PH and the associated progenitor cell accumulation, but the combined CXCR4/CXCR7 antagonism enables baseline maintenance of RVSP, RV/(LV+S) ratio, pulmonary arterial media thickness, as well as perivascular haematopoietic c-kit+ progenitor cell count. Mazzinghi et al. [29] have shown that the CXCL12/CXCR4 interaction is responsible for the recruitment of renal multipotent progenitors, but a CXCL12/CXCR7 interaction is also required for their transendothelial migration. In our study, we determined that CXCR4 was expressed by haematopoietic c-kit+ and endothelial cells and CXCR7 was mainly expressed by endothelial cells. It can be hypothesised that CXCL12 expressed by endothelial and smooth muscle cells acts on CXCR7 expressed by endothelial cells (as paracrine and autocrine secretion) and induces activation of the endothelial cells, and thus, increases adhesion and transendothelial migration of progenitor cells. BM-derived cells were shown to differentiate into α-SMA+ cells in remodelled pulmonary arteries in hypoxic conditions, and selective depletion of circulating mesenchymal precursors with serial intravenous injections of liposomes containing clodronate or of gadolinium chloride prevented pulmonary adventitial remodelling [30], suggesting that these cells may have functional relevance.
Studying the destiny of progenitor cells was not in the scope of this study and deserves further investigations with refined kinetics studies, as well as studies of the mechanism of action of CXCR4, CXCR7 and c-kit antagonists. We cannot exclude that beneficial treatment with CXCR4 and CXCR7 antagonists may be explained not only by the decreased accumulation of haematopoietic progenitor cells in the perivascular area but also by potential effects on the inflammatory response, described in chronic hypoxia, which may promote a progenitor cell's recruitment.
In conclusion, our data demonstrated that mobilisation and homing of haematopoietic c-kit+ progenitor cells in remodelled pulmonary arteries of hypoxic lung were potently reduced by CXCR4 and combined CXCR4 and CXCR7 antagonism. Moreover, these effects on c-kit+ cell accumulation were associated with prevention of PH and pulmonary vascular remodelling. This is the first study demonstrating that combination of AMD3100 and CCX771 reduces the number of perivascular c-kit+ progenitors in hypoxia-induced PH. This study offers important pathophysiological insights into the role of haematopoietic c-kit+ progenitor cells in adult chronic hypoxia-induced vascular remodelling and may have important implications for human PH.
Acknowledgments
We would like to thank ChemoCentryx Inc. (Mountain View, CA, USA) for the kind gift of CXCR7 antagonist CCX771. We also thank A. Huertas (Faculté de médecine, Université Paris-Sud, Kremlin-Bicêtre, INSERM U999, Hypertension Artérielle Pulmonaire, Physiopathologie et Innovation Thérapeutique, Centre Chirurgical Marie Lannelongue, Le Plessis-Robinson and Centre National de Référence de l'Hypertension Artérielle Pulmonaire, Service de Pneumologie et Réanimation Respiratoire, Hôpital Antoine Béclère, AP-HP, Clamart, France) for critical reading of the manuscript and the Plate-forme d'Imagerie Cellulaire Pitié Salpêtrière for confocal imaging.
Footnotes
Support Statement
This study has been supported in part by grants from Ministère de l’Enseignement Supérieur et de la Recherche (GIS-HTAP) and by Legs Poix, Chancellerie des Universités de Paris. N. Gambaryan is supported by a grant from Société de Pneumologie de Langue Française. F. Perros is supported by a Long-Term Fellowship grant of the European Respiratory Society (LTRF 171). D. Montani is supported by a grant from Association HTAPfrance.
Statement of Interest
Statements of interest for D. Montani, G. Simonneau and M. Humbert can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received March 23, 2010.
- Accepted September 18, 2010.
- ©ERS 2011
REFERENCES










