





Abstract
Rhinovirus (RV) infections are the major cause of asthma exacerbations, the major cause of morbidity and mortality in asthma. MUC5AC is the major mucin produced by bronchial epithelial cells. Whether RV infection upregulates MUC5AC in vivo is unknown and the molecular mechanisms involved are incompletely understood.
We investigated RV induction of MUC5AC in vivo and in vitro to identify targets for development of new therapies for asthma exacerbations.
RV infection increased MUC5AC release in normal and asthmatic volunteers experimentally infected with RV-16, and in asthmatic, but not normal, subjects, this was related to virus load. Bronchial epithelial cells were confirmed a source of MUC5AC in vivo. RV induction of MUC5AC in bronchial epithelial cells in vitro occurred via nuclear factor-κB-dependent induction of matrix metalloproteinase-mediated transforming growth factor-α release, thereby activating an epidermal growth factor receptor-dependent cascade culminating, via mitogen-activated protein kinase activation, in specificity protein-1 transactivation of the MUC5AC promoter.
RV induction of MUC5AC may be an important mechanism in RV-induced asthma exacerbations in vivo. Revealing the complex serial signalling cascade involved identifies targets for development of pharmacologic intervention to treat mucus hypersecretion in RV-induced illness.
- Asthma
- chronic obstructive pulmonary disease
- epidermal growth factor receptor
- MUC5AC
- nuclear factor-κB
- rhinovirus
Acute exacerbations are the major cause of morbidity, mortality and healthcare costs in asthma. Exacerbations continue to occur despite the availability of prophylactic medication 1. Therapies better able to reduce the impact of acute exacerbations are needed.
Increased production and secretion of mucus is associated with acute exacerbations, accelerated decline in lung function and fatal asthma 2–4. 21 different mucin genes have been identified. Of these, MUC5AC and MUC5B are the major respiratory mucins produced by bronchial epithelium and submucosal glands, respectively 5, 6. MUC5AC is the predominant mucin in mild-to-moderate asthma 7 and increases in both MUC5AC and MUC5B proteins have been observed in fatal asthma 4. MUC5AC is induced in respiratory epithelial cells by a wide variety of stimuli implicated in the pathogenesis of asthma, including cytokines (interleukin (IL)-9 and IL-13), neutrophil elastase, epidermal growth factor receptor (EGFR) ligands and air pollutants 5; however, the importance and mechanisms of mucin induction in asthma exacerbations has not been extensively investigated.
Rhinoviruses (RV) are associated with the majority of asthma exacerbations 8. Human experimental RV infections have been used to investigate exacerbation pathogenesis, with infection increasing markers of eosinophil activation, IL-8 and neutrophilia 9. Only one study has investigated RV induction of mucus secretion in humans 10; however, this study did not evaluate specific mucin gene expression or protein release, relate virus load to mucin secretion or evaluate the molecular signalling pathways involved.
The primary target of RV is the bronchial epithelial cell 11. One recent report investigated RV induction of MUC5AC in tracheal epithelial cells and reported nuclear factor (NF)-κB, mitogen-activated protein kinase kinase (MEK) and Src to be involved 12. In contrast, most other reports investigating induction of MUC5AC by other stimuli in respiratory epithelial cells implicate EGFR and downstream pathways 13–15. Therefore, the full mechanistic pathways involved in RV induction of MUC5AC remain unclear.
We hypothesised that RV infection would induce MUC5AC secretion in vivo and, having confirmed this, investigated the detailed molecular regulation of RV induction of MUC5AC in vitro. Our findings provide targets for the development of new therapies for mucus hypersecretion in RV-induced illness.
MATERIALS AND METHODS
An experimental model for RV-induced acute exacerbations of asthma
The clinical model, sampling and analysis are described in detail previously 16. Briefly, experimental infections were induced in RV-16-seronegative, atopic asthmatic and normal nonatopic adult subjects by inoculating a 10,000 tissue culture infectious dose (TCID50) per mL dose on day 0 by nasal spray. The study was approved by St Mary's NHS Trust ethics committee (London, UK) and all subjects gave informed consent. Bronchoalveolar lavage (BAL) and bronchial biopsies were taken at baseline ∼2 weeks prior to infection, on day 4 after virus inoculation during acute infection and at convalescence (6 weeks after infection). BAL was assayed by ELISA for MUC5AC protein as described later. Peak virus load during acute infection was determined in daily nasal lavage samples taken for 8 days following inoculation by quantitative (q)PCR as described previously 16.
Immunostaining of MUC5AC in bronchial biopsies
Biopsies were fixed and set in paraffin wax. Sections were stained with mouse anti-human MUC5AC (1:2000 (v/v); Santa Cruz Biotechnology, Santa Cruz, CA, USA) and biotinylated horse anti-mouse antibody (Ab) and counterstained with haematoxylin.
Cell and viral culture
NCI-H292 cells were cultured in RPMI-1640 supplemented with 10% (v/v) fetal calf serum (Invitrogen, Paisley, UK). RV stocks were grown in HeLa cells 17. Viruses were titrated on HeLa cells to ascertain their TCID50 18. The identities of all RVs were confirmed by neutralisation using serotype-specific Ab (ATCC, Manassas, VA, USA). Ultraviolet (UV) inactivation was performed and filtered virus produced by passing RV stocks through a 30 kDa membrane (Millipore, Watford, UK) 17.
Infection of cells with RV
NCI-H292 cells were cultured for 24 h before being serum-starved for 24 h. Cells were infected at a multiplicity of infection (MOI) of 1 (unless otherwise stated) for 1 h.
For inhibition studies, actinomycin D, AG1478, PD98059, U0126, mithramycin A, CAPE, cycloheximide and GM6001 were purchased from Calbiochem (Darmstadt, Germany). AS602868 was kindly provided by I. Adcock (Imperial College London, London, UK). Cells were pre-treated for 1–2 h before infection.
For the MUC5AC promoter studies, NCI-H292 cells were transfected with MUC5AC promoter constructs 15, including 0.4 μg MUC5AC-construct, 0.2 μg pCMVSPORT-βgal (Invitrogen) and 0.4 μg poly(rI:C) or poly(dI:C) as control (Sigma–Aldrich, Poole, UK). Luciferase levels were assessed and normalised to β-galactosidase levels (Promega, Southampton, UK) 15.
ELISA to evaluate MUC5AC protein
NCI-H292 culture supernatants and diluted BAL (1:100 (v/v) in carbonate-bicarbonate buffer) were assayed for MUC5AC protein by ELISA 15.
RNA extraction, reverse transcription and real-time qPCR
RNA extraction, reverse transcription and qPCR analysis of MUC5AC gene expression was performed as previously described 15.
Site-directed mutagenesis of MUC5AC -324 promoter construct
Site-directed mutagenesis of the NF-κB and interferon regulatory factor (IRF) transcription factor binding sites was carried out as previously described 15. The mutations introduced were: NF-κB, GGGGAGGACCCCT to TTTTAGGACCCCT; IRF, TCACTTCTGG to TCACGGGACC (mutated bases underscored).
Western blotting for EGFR phosphorylation
Cells were infected with RV-16, or transfected with 0.4 μg poly(rI:C) or poly(dI:C). Cells were harvested, and proteins resolved and analysed by immunoblotting with mouse anti-phospho-EGFR or sheep anti-EGFR (1:1000 (v/v); Upstate, Watford, UK) and horseradish peroxidase-conjugated secondary antibodies.
ELISA to quantify extracellular signal-regulated kinases 1/2 phosphorylation
At 3, 6 and 24 h post-infection, proteins were extracted using Cell Extraction Buffer (Biosource, Paisley, UK) including protease inhibitors (Pierce Biotechnology, Cramlington, UK). Total and phospho-specific extracellular signal-regulated kinase (ERK)1/2 were quantified using specific ELISAs (Biosource).
ELISA to quantify transforming growth factor-α release
At 8 h post-infection, supernatants were harvested and assayed for transforming growth factor (TGF)-α release using a human TGF-α Quantikine ELISA (R&D Systems, Abingdon, UK).
Confocal microscopy for RV infection and p65 translocation
At 6 h post-infection, cells were fixed, permeabilised and incubated with a rabbit anti-RV 3C protease Ab (1:200 (v/v); provided by J. Gern, University of Wisconsin, Madison, WI, USA 19) and mouse anti-p65 Ab (1:500 (v/v); Santa Cruz Biotechnology). These were detected using goat anti-mouse AlexaFlour546 (1:200 (v/v)) and anti-rabbit AlexaFlour488 (1:200 (v/v)) Ab (Invitrogen). Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
Statistical analysis
Data are presented as mean±sem. All data were analysed using ANOVA and Bonferroni’s multiple-comparison, post hoc test. Correlations between MUC5AC concentrations and virus load were examined using Spearman’s rank correlation. Data were accepted as significantly different when p<0.05.
RESULTS
RV induction of MUC5AC protein secretion in vivo and relation to virus load
Nine atopic asthmatic and 15 nonatopic normal volunteers were experimentally infected with RV-16 as described previously 16. MUC5AC protein was quantified in BAL harvested prior to (baseline), and 4 days and 6 weeks after infection. In addition, nasal lavage samples were taken to determine peak virus load during the infection 16.
RV-infection led to a significant increase in BAL MUC5AC protein between the baseline and acute infection phases (p = 0.033), which was resolved by 6 weeks (p = 0.002; fig. 1a). Asthmatic patients had greater mean levels of MUC5AC BAL protein at each time point compared with normal subjects; however, these differences were not statistically significant (fig. 1b).

MUC5AC is induced by rhinovirus (RV) in vivo, correlating with virus load and produced by bronchial epithelial cells. Bronchoalveolar lavage (BAL) and nasal lavage were taken from 15 normal and nine asthmatic subjects at baseline, and 4 days and 6 weeks post-infection. MUC5AC was quantified in the BAL by ELISA. RV load was quantified by quantitative PCR in nasal lavage. a) Quantification of MUC5AC protein secreted into BAL for all patients. Boxes represent median and interquartile range, and whiskers respresent 95% CI. ○: outliers (1.5–2-times the interquartile range); •: extremes (>2-times the interquartile range). #: p = 0.033; ¶: p = 0.002. b) Quantification of MUC5AC protein secreted into BAL for normal and asthmatic patients. +: p = 0.32; §: p = 0.44; ƒ: p = 0.26; ##: p = 0.14; ¶¶: p = 0.047; ++: p = 0.1; §§: p = 0.066. c) Correlation between peak virus load and BAL MUC5AC at day 4 in asthmatic (•; p = 0.02, r = 0.750) and normal (○; p = 0.379, r = 0.245) subjects. —: correlation for asthmatic subjects. d) Bronchial biopsies were taken at baseline and 4 days post-infection. Biopsies were stained for MUC5AC (brown) and counterstained with haematoxylin (blue). Scale bars = 50 μm. Representative immunohistochemistry images from a normal subject at 1) baseline and 2) day 4, and at 3) baseline and 4) day 4 in an asthmatic patient.
In asthmatic patients alone, MUC5AC levels in BAL were highly correlated with virus load (r = 0.750, p = 0.02; fig. 1c), indicating induction was related to severity of infection. No significant relationship was observed for normal subjects.
Bronchial epithelial cells are a source of MUC5AC in vivo
Bronchial biopsies taken at baseline and 4 days post-infection 16 were stained for MUC5AC protein expression. In both normal (fig. 1d, upper panels) and asthmatic (fig. 1d, lower panels) subjects, epithelial cells were identified as MUC5AC-producing cells. Quantitative analysis could not be performed due to loss of epithelium in many of the asthmatic subjects and, therefore, while we were able to identify RV-induced release of MUC5AC (figs 1a and b) we were unable to definitively demonstrate an increase in production. However, these data do demonstrate that bronchial epithelial cells are a source of MUC5AC protein in the human lung in vivo.
RV stimulates de novo MUC5AC synthesis and secretion in respiratory epithelial cells in vitro
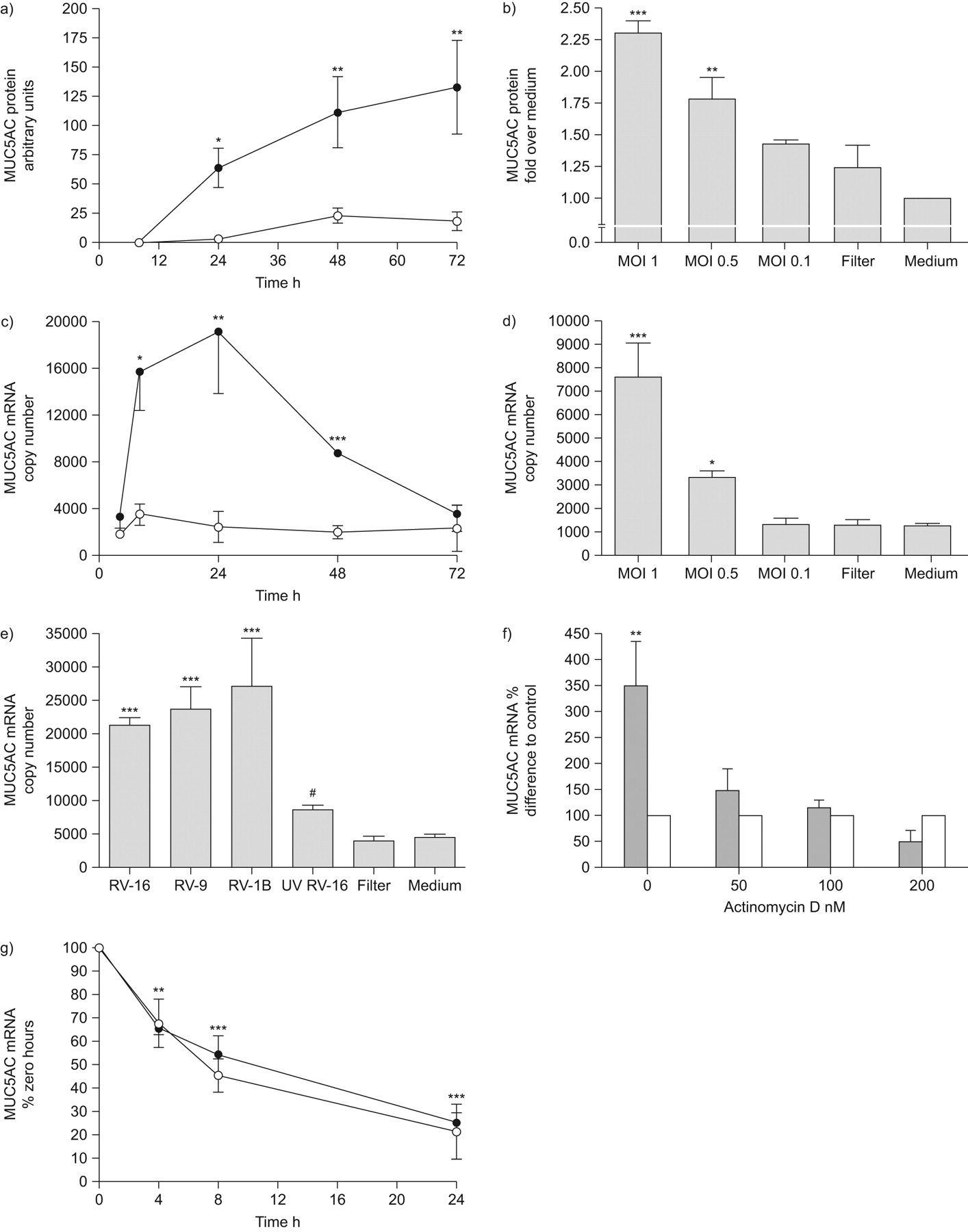
Having found RV induction of MUC5AC in vivo, we then investigated mechanisms in vitro. We infected the bronchial epithelial cell line NCI-H292 with RV-16 and observed a time-dependent increase in MUC5AC protein compared to uninfected control cells (fig. 2a). This induction was dose-dependent, with significant induction of MUC5AC protein by RV-16 at an MOI of 1 and 0.5, but not 0.1 (fig. 2b).

Rhinovirus (RV) infection leads to time-, dose- and replication-dependent increases in de novo MUC5AC expression. a) Cells were infected with RV-16 and MUC5AC protein quantified by ELISA at 8, 24, 48 and 72 h post-infection (n = 5). •: RV-16; ○: control. b) Cells were infected with RV-16 at multiplicity of infection (MOI) of 0.1, 0.5 and 1, or equivalent volume of filtered RV-16, and MUC5AC protein quantified by ELISA at 24 h post-infection (n = 3). c) Cells were infected as in a) and MUC5AC mRNA harvested and quantified by quantitative (q)PCR (n = 4). •: RV-16; ○: control. d) Cells were infected as in b), and MUC5AC mRNA was harvested and quantified by qPCR 8 h post-infection (n = 4). e) Cells were infected with RV-16, RV-1B, RV-9, ultraviolet-inactivated RV-16 or an equivalent volume of filtered RV-16, and MUC5AC mRNA was quantified by qPCR 8 h post-infection (n = 5). *: p<0.05 comapred to uninfected control cells; **: p<0.01 compared to uninfected control cells; ***: p<0.001 compared to uninfected control cells; #: p<0.001 compared to RV-16-infected cells. f) Cells were pre-treated with actinomycin D at doses indicated and infected with RV-16. MUC5AC mRNA was quantified by qPCR 8 h post-infection (n = 4). ▓: RV-16; □: medium. g) Cells were infected with RV-16 and treated with 100 nM actinomycin D 16 h later. MUC5AC mRNA was harvested and quantified by qPCR at 0, 4, 8 and 24 h post-actinomycin D (n = 4). Data are presented as mean±sem. •: RV-16; ○: control. **: p<0.01 compared to time 0; ***: p<0.001 compared to time 0.
To determine whether RV increased MUC5AC transcription, we assessed MUC5AC mRNA expression. This was significantly increased 8–48 h post-infection, peaking at 24 h, but had returned to baseline by 72 h (fig. 2c). Induction of mRNA expression was dose-dependent (fig. 2d).
As the RV family contains both major and minor serotypes (which use different receptors to infect), we demonstrated that RV-16, RV-9 (both major group) and RV-1B (minor group) significantly increased MUC5AC expression (fig. 2e), indicating that induction was neither serotype- nor receptor-restricted. In addition, we confirmed induction was virus-specific, as virus-free inocula and UV-inactivated RV-16 failed to significantly induce MUC5AC expression (fig. 2e).
We next investigated whether the increased MUC5AC expression was due to increased de novo transcription, using actinomycin D to inhibit new mRNA synthesis and suppress induction of MUC5AC mRNA expression (fig. 2f), confirming increased expression was due to increased transcription. Additionally, we monitored MUC5AC mRNA over time and observed no difference in degradation rates in the presence of RV-16 (fig. 2g), confirming that RV-16 infection did not alter MUC5AC mRNA degradation.
RV induces MUC5AC expression via an EGFR–MEK/ERK–specificity protein-1 signalling cascade
Previous studies report EGFR activation is required for induction of MUC5AC expression by a wide range of mediators 13–15. Therefore, we investigated this for RV-induced MUC5AC expression.
AG1478, a specific inhibitor of EGFR phosphorylation and activation, resulted in dose-dependent and complete inhibition of MUC5AC expression (fig. 3a). We next confirmed that RV infection activated EGFR using phospho-specific Western blotting (fig. 3b) and also confirmed that poly(rI:C), a frequently used model of virus infection, activated EGFR in the same manner (fig. 3c).
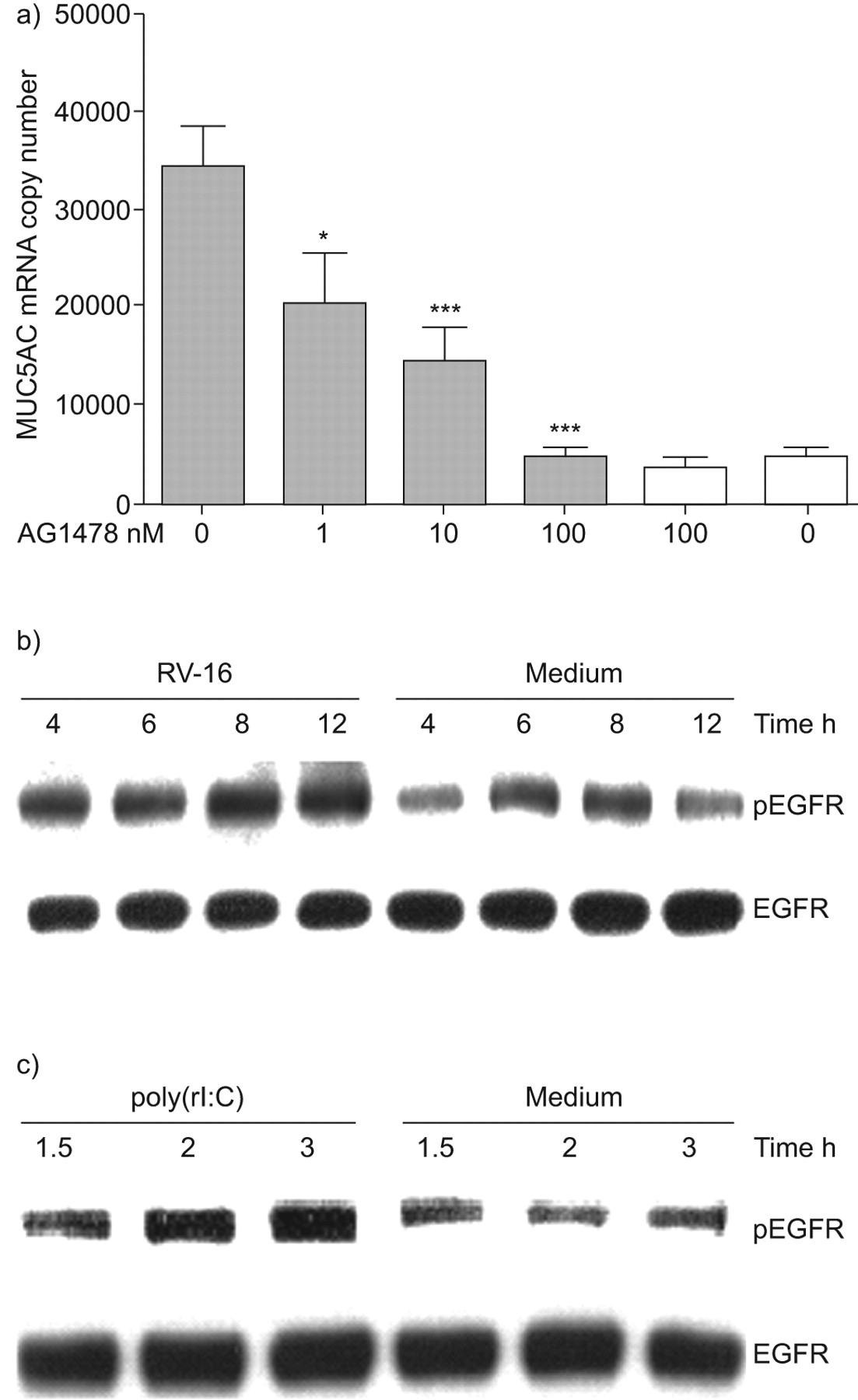
Rhinovirus (RV) induction of MUC5AC requires activation of epidermal growth factor receptor (EGFR). a) Cells were pre-treated with AG1478 at doses indicated and infected with RV-16. MUC5AC mRNA was harvested and quantified by quantitative (q)PCR 8 h post-infection (n = 5). Data are presented as mean±sem. ▓: RV-16; □: uninfected. *: p<0.05 compared to control infected cells; ***: p<0.001 compared to control infected cells. b) Cells were infected with RV-16 and proteins harvested 4, 6, 8 and 12 h post-infection. Total EGFR and phospho-specific (p)EGFR were detected by Western blotting. c) Cells were stimulated with poly(rI:C) or poly(dI:C) as control, and proteins harvested 1.5, 2 and 3 h later. Total EGFR and pEGFR was detected by Western blotting. Western blots are representative of three experiments.
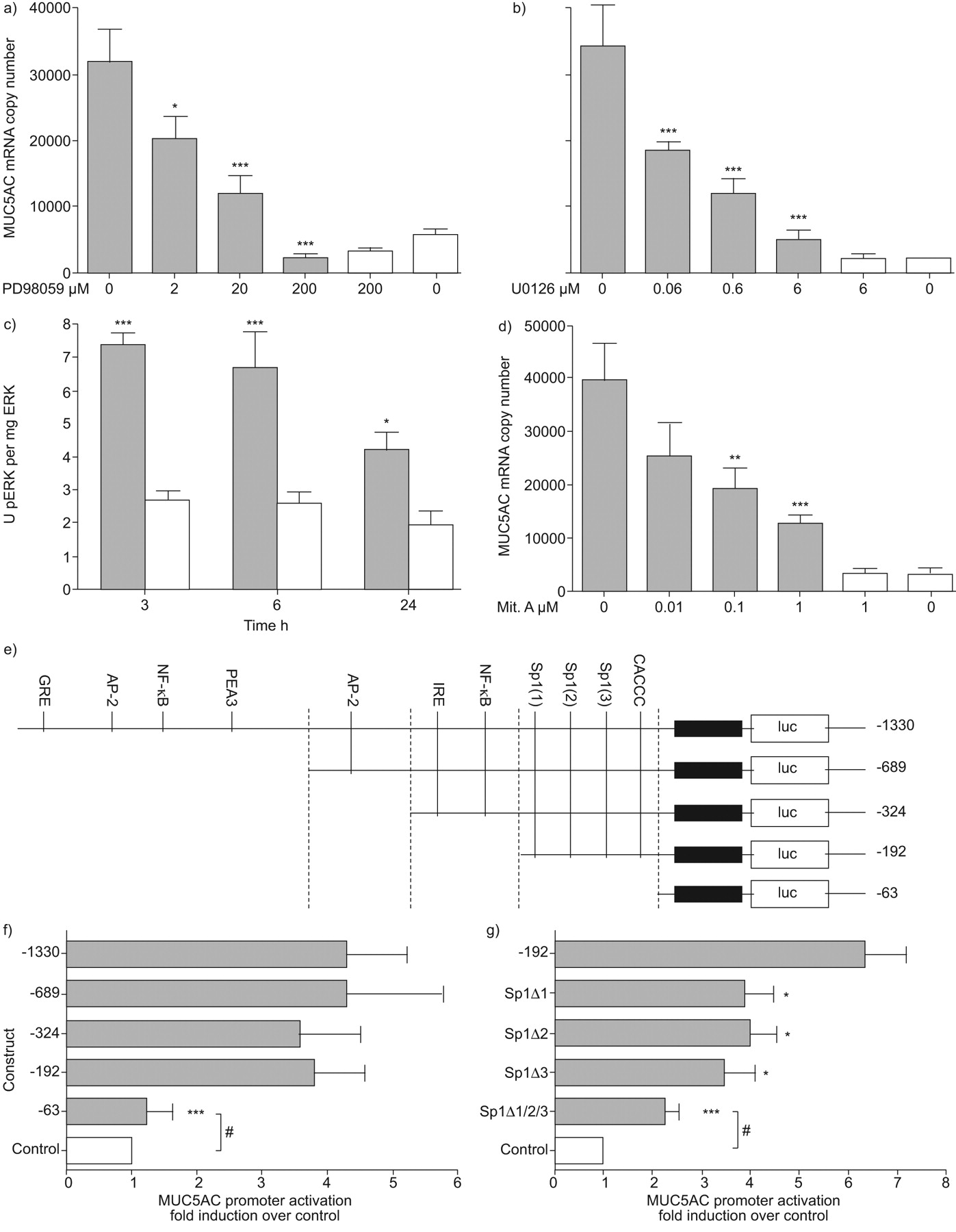
We then investigated the signalling pathways downstream of EGFR. PD98059, an inhibitor of MEK activation, led to a dose-responsive and complete inhibition of MUC5AC mRNA expression (fig. 4a), as did a second MEK-specific inhibitor, U0126 (fig. 4b). We confirmed that RV infection induced ERK phosphorylation from 3–24 h (fig. 4c) and that mithramycin A, an inhibitor of specificity protein (Sp)1 binding, resulted in a dose-responsive inhibition of MUC5AC expression (fig. 4d). To confirm the requirement for Sp1, we transfected NCI-H292 cells with MUC5AC promoter–luciferase constructs (fig. 4e) 15 and observed that serial truncation of the promoter from -1330 to -192 bp did not alter induction of the MUC5AC promoter by poly(rI:C) (fig. 4f); however, when the promoter was further truncated to the -63 bp fragment, thus removing the Sp1 sites, induction was abolished. Finally, we mutated the three Sp1 sites within the -192 bp promoter fragment, confirming that all three sites were active, as on deletion of all Sp1 sites there was no longer significant induction (fig. 4g).

Rhinovirus (RV) induction of MUC5AC requires mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) and specificity protein (Sp)1 transactivation of the MUC5AC promoter. a) Cells were pre-treated with PD98059 at doses indicated and infected with RV-16. MUC5AC mRNA was quantified by quantitative (q)PCR 8 h post-infection (n = 4). *: p<0.05 compared to uninhibited infected cells; ***: p<0.001 compared to uninhibited infected cells. b) As a), but cells pre-treated with U0126 as indicated (n = 4). ***: p<0.001 compared to uninhibited infected cells. c) Cells were infected with RV-16 and total ERK or phospho-ERK (pERK) quantified by ELISA at 3, 6 and 24 h post-infection (n = 4). *: p<0.05 compared to uninfected control cells; ***: p<0.001 compared to uninfected control cells. d) As a) and b), but cells pre-treated with mithramycin A (Mit. A) as indicated (n = 5). **: p<0.01 compared to uninhibited infected cells; ***: p<0.001 compared to uninhibited infected cells. e) Map of cloned MUC5AC promoter, indicating the five truncations and putative transcription factor binding sites. GRE: granulocyte colony-stimulating factor-responsive element; AP-2: activating protein-2; NF-κB: nuclear factor-κB; IRE: interferon-responsive element. f) Cells were transfected with the five constructs, alongside poly(rI:C) or poly(dI:C) as control. Luciferase (luc) expression was quantified 48 h post-infection (n = 5). ***: p<0.001 compared to -1330 construct. g) Cells were transfected with -192 construct containing mutated Sp1 binding sites alongside poly(rI:C), or poly(dI:C) as control. Luc expression was quantified 48 h post-infection (n = 5). *: p<0.05 compared to -192 construct; ***: p<0.001 -192 constructs; #: not significant. Data are presented as mean±sem. ▓: RV-16; □: uninfected.
These data demonstrate that RV induction of MUC5AC expression requires activation of the EGFR, followed by activation of MEK and ERK, culminating in transactivation of the MUC5AC promoter by Sp1 binding to three specific sites within the proximal promoter.
RV induction of MUC5AC requires NF-κB mediated induction of TGF-α release upstream of EGFR activation
With the identification that EGFR was required and knowing that EGFR ligands are cleaved from precursors by matrix metalloproteinases (MMP) that may be induced by NF-κB, which is implicated in RV-induced inflammation 17, we next investigated the role NF-κB in RV induction of MUC5AC.
Two pharmacologically distinct inhibitors of NF-κB activation, caffeic acid phenethyl ester (CAPE), an inhibitor of p65 nuclear translocation (fig. 5a) and AS602868, an inhibitor of inhibitor of κB kinase (IKK)-β (fig. 5b) resulted in a dose-responsive inhibition of MUC5AC induction. To confirm that RV infection caused activation of NF-κB, using confocal microscopy, we stained cells for RV 3C protease, a nonstructural protein expressed only in actively infected cells 19, the p65 subunit of NF-κB and DAPI. We observed NF-κB nuclear translocation (red) only in cells infected with RV (green), while uninfected cells retained p65 in their cytosol (fig. 5c).

Rhinovirus (RV) induction of MUC5AC requires nuclear factor-κB activation. a) Cells were pre-treated with caffeic acid phenethyl ester (CAPE) at doses indicated and infected with RV-16. MUC5AC mRNA was quantified by quantitative PCR 8 h post-infection (n = 4). b) As a), but cells pre-treated with AS602868 as indicated (n = 4). ▓: RV-16; □: uninfected. Data are presented as mean±sem. *: p<0.05 compared to control infected cells; ***: p<0.001 compared to control infected cells. c) Cells were infected with RV-16 before RV 3C protease and p65 were detected and visualised by confocal microscopy 6 h post-infection. Arrows indicate co-localised 3C protease and p65 nuclear translocation. Confocal image is representative of three experiments. Scale bars = 12.5 μm.
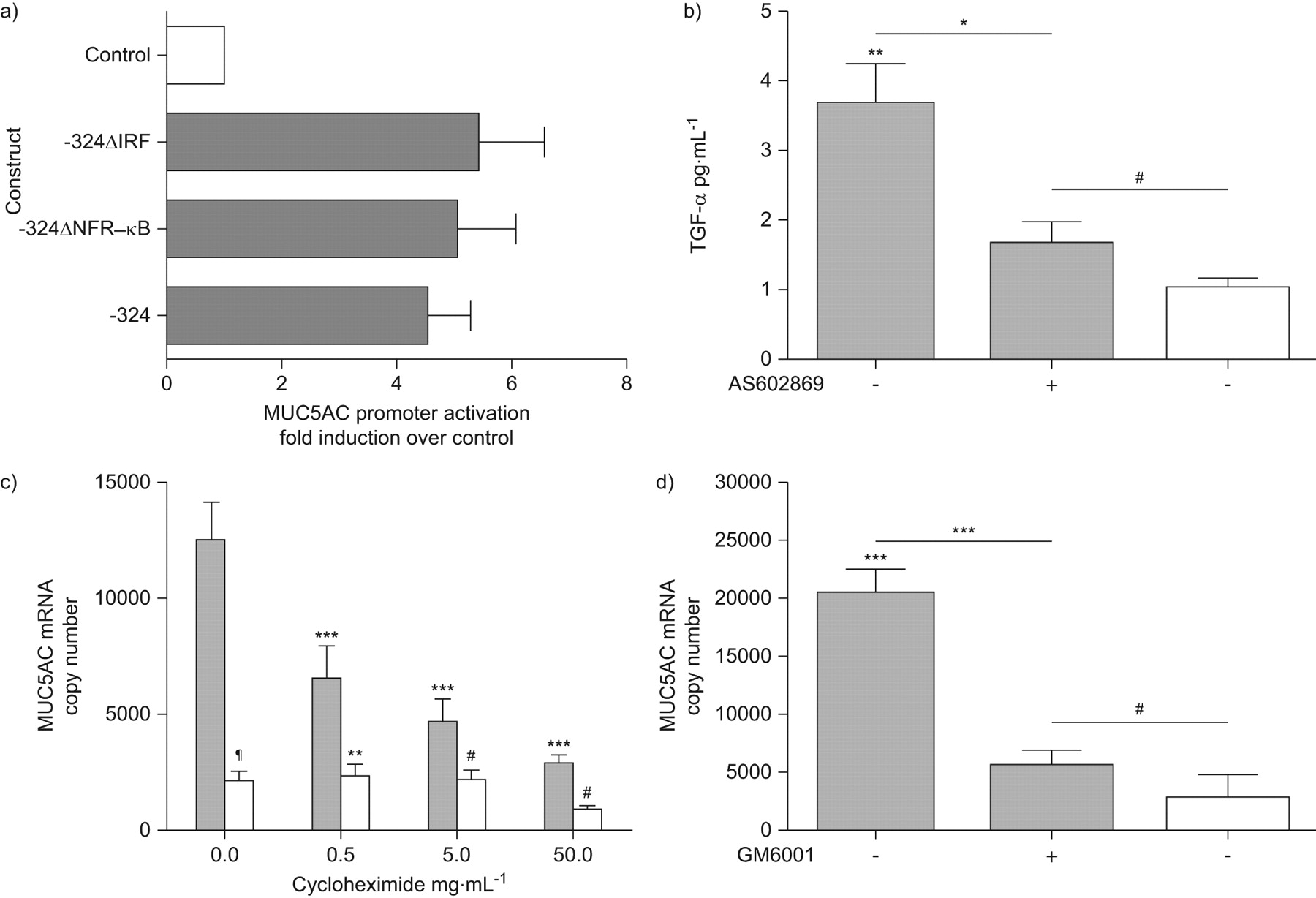
Virus infections induce both NF-κB and interferon regulatory factors and the MUC5AC promoter contained binding sites for both (fig. 4e). Site-directed mutagenesis of these sites did not alter promoter activity of the -324 construct (fig. 6a).

Rhinovirus (RV) induction of MUC5AC requires a nuclear factor (NF)-κB-dependent, matrix metalloprotease-mediated release of transforming growth factor (TGF)-α. a) Cells were transfected with wild type or -324 constructs containing mutated NF-κB and interferon regulatory factor (IRF) binding sites, alongside poly(rI:C), or poly(dI:C) as control. Luciferase expression was quantified 48 h post-infection (n = 4). ▒: poly(rI:C); □: control. b) Cells were pre-treated with 1×10−6 M AS602868 and infected with RV-16. TGF-α release was quantified 6 h post-infection (n = 3). *: p<0.05 compared to control cells; **: p<0.01 compared to control cells; #: not signficant. c) Cells were pre-treated with cycloheximide at doses indicated and infected with RV-16. MUC5AC mRNA was quantified 8 h post-infection (n = 3). **: p<0.01 comparing infected and control cells at each concentration of cycloheximide; ***: p<0.001 compared to control infected cells; #: not significant; ¶: p<0.001 comparing infected and control cells at each concentration of cycloheximide. d) Cells were pre-treated with 4×10−5 M GM6001 and infected with RV-16. MUC5AC mRNA was quantified 8 h post-infection (n = 3). ***: p<0.001 compared to control cells; #: not significant. Data are presented as mean±sem. ▓: RV-16; □: uninfected.
Having demonstrated that NF-κB did not transactivate the MUC5AC promoter directly, we investigated the role of NF-κB in activation of EGFR ligands. Pro-transforming growth-factor (TGF)-α cleavage, activation and release from the cell surface has previously been demonstrated to be upstream of MUC5AC expression 20. Therefore, we investigated whether RV infection induced the release of TGF-α and whether this was NF-κB-dependent using AS602868. Infection of NCI-H292 cells with RV-16 resulted in significant induction of TGF-α release into supernatants, which was significantly blocked by NF-κB inhibition (fig. 6b).
Next, we sought to confirm that RV induction of MUC5AC expression required the synthesis of an intermediate protein. Treatment of cells with cycloheximide to prevent de novo protein synthesis significantly inhibited MUC5AC gene expression (fig. 6c), confirming requirement of a newly synthesised intermediate protein in this process.
As MMPs are known to be induced by NF-κB and to cleave pro-TGF-α to active TGF-α 21, we investigated the involvement of MMPs using the inhibitor GM6001 (fig. 6d) and observed that GM6001 significantly inhibited RV induction of MUC5AC mRNA.
These data demonstrate that RV induction of MUC5AC expression requires NF-κB activation in addition to the EGFR–MEK/ERK–Sp1 pathway. However, NF-κB does not directly transactivate the MUC5AC promoter, but activates expression and translation of intermediate MMPs that induce TGF-α release upstream of EGFR activation.
RV induction of MUC5AC protein requires the same key signalling events
Finally, having defined in detail the mechanisms involved in RV induced MUC5AC gene expression, we wished to confirm our key findings for MUC5AC protein release.
We repeated our experiments using the inhibitor of NF-κB (CAPE), the inhibitor of MMP (GM6001), the inhibitor of the EGFR (AG1478) and the inhibitors of MEK activation (PD98059 and U0126), and demonstrated a statistically significant reduction in RV-induced MUC5AC protein release with all inhibitors (fig. 7).

Rhinovirus (RV) induction of MUC5AC protein release requires activation of nuclear factor-κB, matrix metalloprotease, epidermal growth factor receptor and mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathways a) Cells were pre-treated with caffeic acid phenethyl ester (CAPE) at doses indicated and infected with RV-16. MUC5AC protein was quantified by ELISA 24 h post-infection (n = 7). b) As a), but cells pre-treated with GM6001 as indicated (n = 3). c) As a), but cells pre-treated with AG1478 as indicated (n = 4). d) As a), but cells pre-treated with PD98059 as indicated (n = 4). e) As a), but cells pre-treated with U0126 as indicated (n = 3). Data are presented as mean±sem. ▓: RV-16; □: uninfected. *: p<0.05 compared to uninhibited infected cells; **: p<0.01 compared to uninhibited infected cells; ***: p<0.001 compared to uninhibited infected cells.
These data confirmed the signalling pathway of RV induction of NF-κB to activate MMPs and a subsequent EGFR–MEK/ERK pathway leading to MUC5AC synthesis and secretion.
DISCUSSION
RVs are the major cause of asthma exacerbations and mucus secretion is important in exacerbation pathogenesis; however, RV induction of MUC5AC has not been demonstrated in humans and the molecular mechanisms regulating RV induction of MUC5AC are poorly understood.
We have demonstrated that MUC5AC release into BAL is increased by RV infection in vivo and, in asthmatic subjects only, BAL MUC5AC levels correlated with peak virus load. It is likely that this correlation is related to the impaired innate antiviral immune response to RV infection recently reported in bronchial epithelial cells in asthma 11.
We next confirmed bronchial epithelial cells as a source of MUC5AC in vivo and observed that RV infection stimulated de novo MUC5AC gene expression and protein secretion from respiratory epithelial cells in vitro. This was RV serotype- and receptor-independent and replication-dependent. Next, we identified activation of the EGFR–MEK/ERK signalling pathway, culminating in Sp1 binding the proximal MUC5AC promoter. In addition, we identified a second signalling cascade upstream of EGFR activation: RV induction of MUC5AC was NF-κB-dependent; however, NF-κB did not directly transactivate the promoter. Instead, NF-κB was required for MMP-dependent release of the EGFR ligand TGF-α 14, thus completing a complex signalling cascade (fig. 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Molecular mechanisms of rhinovirus (RV) induction of MUC5AC synthesis and secretion. RV infects respiratory epithelial cells causing activation and nuclear translocation of nuclear factor (NF)-κB. This causes transcription and translation of matrix metalloproteases (MMPs) that cleave pro-transforming growth factor (TGF)-α and active TGF-α is released from the cell surface. The active TGF-α then binds to and activates the epidermal growth factor receptor (EGFR) present on the surface epithelium in a para/autocrine mechanism. Phosphorylation of the intracellular domain of EGFR activates a cellular signalling cascade including mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinase (ERK) activation and culminates in specificity protein (Sp)1 transactivation of the MUC5AC promoter. This drives de novo transcription of the MUC5AC gene, increasing MUC5AC mRNA quantities and, ultimately, resulting in secretion of MUC5AC protein.
A recent publication has described RV-14 mediated upregulation of MUC5AC via Src/MEK/NF-κB 12. However, those authors overlooked the role of the EGFR and implied that NF-κB induced MUC5AC release directly. We have demonstrated that NF-κB is involved indirectly via increasing transcription of an intermediate MMP. Another recent publication also identified involvement of the EGFR–ERK pathway in RV/double-stranded RNA induction of MUC5AC. In that case, initiated by Toll-like receptor (TLR)-3 recognition of the poly(rI:C) used 22. While that paper did not fully characterise the complete pathway described here and did not identify the induction of an intermediate MMP, their identification of upstream TLR-3 signalling increases the range of potential therapeutic targets for development of new approaches to therapy.
This combination of two serially linked signalling cascades, both necessary for induction of MUC5AC, is a novel pathway for mucin induction, so far unique for RV. However, it is possible that studies investigating other stimuli may have overlooked this pathway. We have previously reported that for phorbol 12-myristate 13-acetate (PMA)-induced MUC5AC, an EGFR pathway culminating in Sp1 transactivation of the promoter was required 15 and others have highlighted Sp1 transactivation of MUC2 and MUC5AC promoters 23. We noted at that time that PMA induction of MUC2 in colonic epithelium was also reported to involve EGFR-mediated signalling pathways but, in contrast, culminated in NF-κB activation of the promoter 24. However, those authors did not demonstrate any effect on MUC2 promoter activity when the putative NF-κB site was removed. With our demonstration that both NF-κB and Sp1 are required in the complex RV induction of MUC5AC, it is possible that both are also required for induction of MUC2 and MUC5AC by other stimuli. There are several other reports demonstrating Sp1 mediates activation of MUC2 in intestinal 25 and respiratory epithelium 26; and there are, equally, several reports of a requirement for NF-κB in MUC2 27 as well as MUC5AC expression 28. With the extensive literature establishing the importance of the EGFR pathway (which culminates in Sp1 transactivation of MUC2 and MUC5AC promoters) 13, 14, it is likely that these studies identifying a requirement for NF-κB may be highlighting this pre-EGFR signalling cascade we have identified with RV. This possibility should, therefore, be considered for other secretagogues.
Our findings also have important implications for acute exacerbations of other illnesses, as RVs are reported to be a major cause of acute exacerbations of chronic obstructive pulmonary disease 29 and other respiratory illnesses 30 in which mucus hypersecretion is likely to play an important role.
In conclusion, this study has identified that RV infection induces MUC5AC protein release in vivo and the key molecules involved in a complex serial induction pathway. These data provide targets for the development of novel interventions against MUC5AC hypersecretion in RV-induced exacerbations of airway diseases.
Footnotes
For editorial comments see page 1236.
Statement of Interest
Statements of interest for C.A. Hewson, M.R. Edbrooke and S.L. Johnston can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received February 18, 2010.
- Accepted March 31, 2010.
- ©ERS 2010
REFERENCES










