





Abstract
Epigenetic mechanisms mediate genomic adaption to the environment and epigenetic alterations can contribute to the development of disease phenotypes, as can genetic variants. A variety of molecular mechanisms are involved in epigenetic regulation, including post-transcriptional histone modifications, histone variants, ATP-dependent chromatin remodelling complexes, polycomb/trithorax protein complexes, small and other noncoding RNAs (small interfering RNA and micro RNAs) and DNA methylation. Epigenetic mechanisms have been identified in cancer but very little is known about these effects in complex diseases such as asthma. Epigenetic mechanisms have been found to play a primordial role in T-cell differentiation and novel aspects of asthma and allergy development are now investigated by systematic epigenetic studies. Here we give an introduction to epigenetics, review the existing literature in relation to asthma and asthma-related mechanisms and hypothesise on feasible approaches for the study of epigenetics in asthma in the future.
Epigenetics can be defined as the study of heritable changes of a phenotype, such as the gene expression patterns of a specific cell type that are not caused by changes in the nucleotide sequence of the genetic code itself. Epigenetic mechanisms convey genomic adaption to an environment, thereby ultimately contributing towards the phenotype. Epigenetic phenomena are mediated by a variety of molecular mechanisms, including post-transcriptional histone modifications, histone variants, ATP-dependent chromatin remodelling complexes, polycomb/trithorax protein complexes, small and other noncoding RNAs (including small interfering RNA and micro RNAs (miRNAs)), and DNA methylation 1. Chromatin modulations play a central role in shaping the epigenome and delineate a functional chromatin topology which serves as the platform forming regulatory circuits in all cells. Open (euchromatin) and closed (heterochromatin) chromatin states are controlled by histone modifications and histone composition in close crosstalk with the binding of a myriad of non-histone proteins (fig. 1). The basic building block of chromatin is the nucleosome, which is formed of an octamer of histone proteins containing an H3–H4 tetramer flanked on either side with an H2A–H2B dimer around which 146 base pairs of DNA are spooled. The protruding N-terminal tails of these histones undergo extensive changes caused by various modifications such as acetylation, methylation, phosphorylation and ubiquitylation 2. The combination of different N-terminal modifications and the incorporation of different histone variants that have distinct roles in gene regulation have led to the proposition of a regulatory histone code, which determines, at least partly, the transcriptional potential for a specific gene or a genomic region 3. These diverse molecular mechanisms have all been found to be closely intertwined and stabilise each other to ensure the faithful propagation of an epigenetic state over time and, in particular, through cell division. Nonetheless, epigenetic states are not definitive and changes occur over time in a stochastic manner as well as in response to environmental stimuli.

Schematic representation of euchromatin and heterochromatin. a) In euchromatin, the promoter CpG island ( ) is an accessible nucleosome-free region. RNA polymerase binding to transcription start is possible. Histone hyperacetylation including lysine 9 of histone H3 (H3K9) is simultaneously present with the methylation on lysine 4 of H3 (H3K4). b) In heterochromatin, the nucleosome structure and, therefore, also the DNA is compacted and the region is transcriptionally silent. H3K4 is demethylated, whereas H3K9, H3K27 and H4K20 are methylated like DNA. For simplification, only selected histone modifications associated with the chromatin state are shown.
) is an accessible nucleosome-free region. RNA polymerase binding to transcription start is possible. Histone hyperacetylation including lysine 9 of histone H3 (H3K9) is simultaneously present with the methylation on lysine 4 of H3 (H3K4). b) In heterochromatin, the nucleosome structure and, therefore, also the DNA is compacted and the region is transcriptionally silent. H3K4 is demethylated, whereas H3K9, H3K27 and H4K20 are methylated like DNA. For simplification, only selected histone modifications associated with the chromatin state are shown.
Epigenetic modifications contribute to so many processes that, for the purposes of this review, oversimplifications and large omissions are unavoidable. In this manuscript, we will mainly concentrate on DNA methylation, the best studied form of epigenetic modification and, very briefly, touch on regulation through miRNA, an epigenetic mechanism acting on gene expression in a dynamic and tissue-specific manner.
DNA METHYLATION
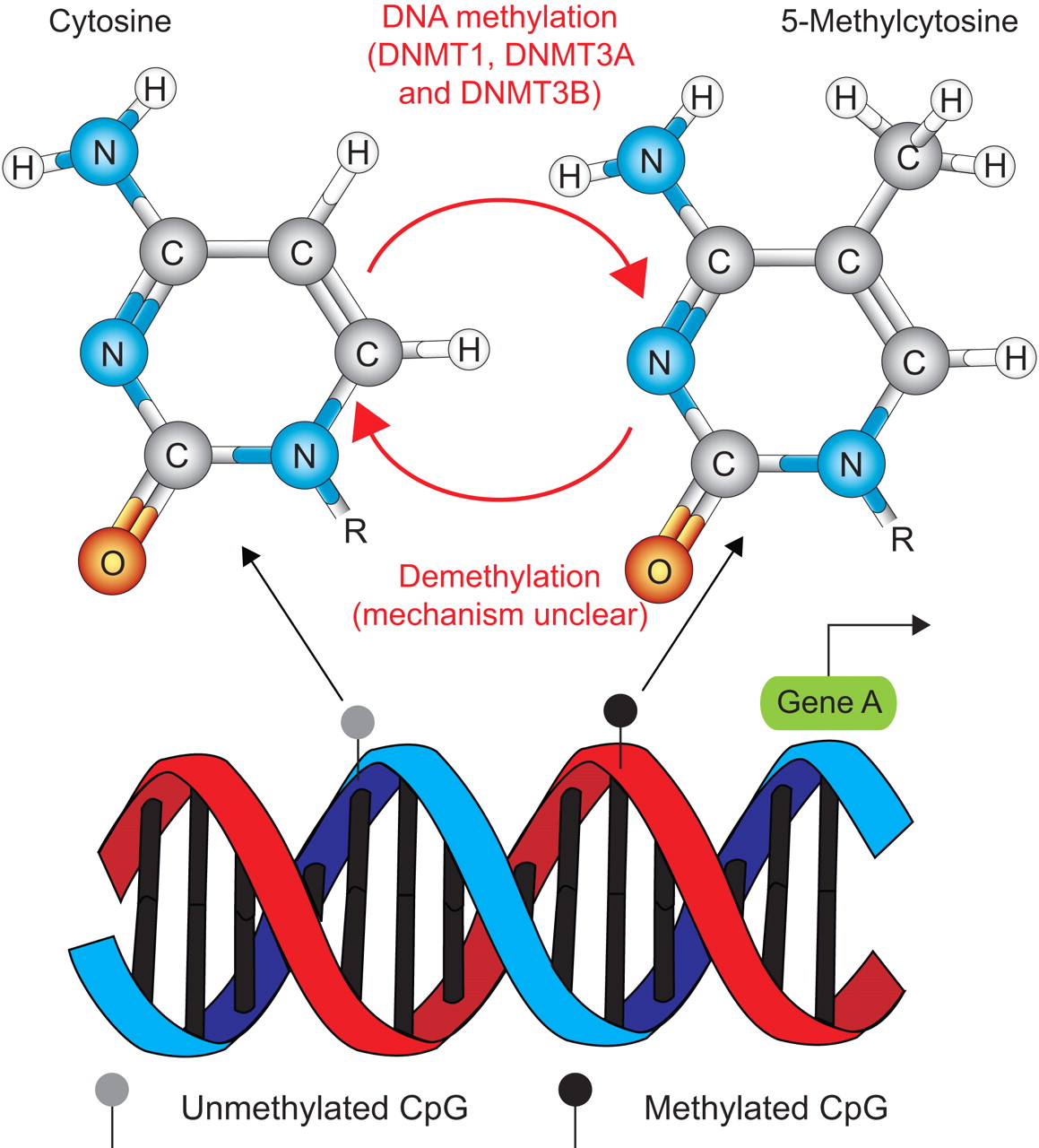
DNA methylation is the only genetically programmed modification of DNA itself that is presently known in mammals. All other epigenetic effects act on proteins interacting with DNA or at the level of RNA interference. This post-replication modification is almost exclusively found on the 5 position of the pyrimidine ring of cytosines in the context of the dinucleotide sequence CpG (fig. 2) 4. 5-Methylcytosine accounts for ∼1% of all bases and the majority (75%) of CpG dinucleotides throughout mammalian genomes are methylated. Owing to their inherent mutagenicity, CpGs are under-represented in the genome, but relatively CpG-rich clusters of approximately 1–4 kb in length, so-called CpG islands, are found in the promoter region and first exons of many genes. In contrast to single CpGs, they are mostly nonmethylated, corresponding to open chromatin structure and a potentially active state of transcription 5. About three-quarters of transcription start sites and 88% of active promoters are associated with CpG-rich sequences and might be regulated by DNA methylation.
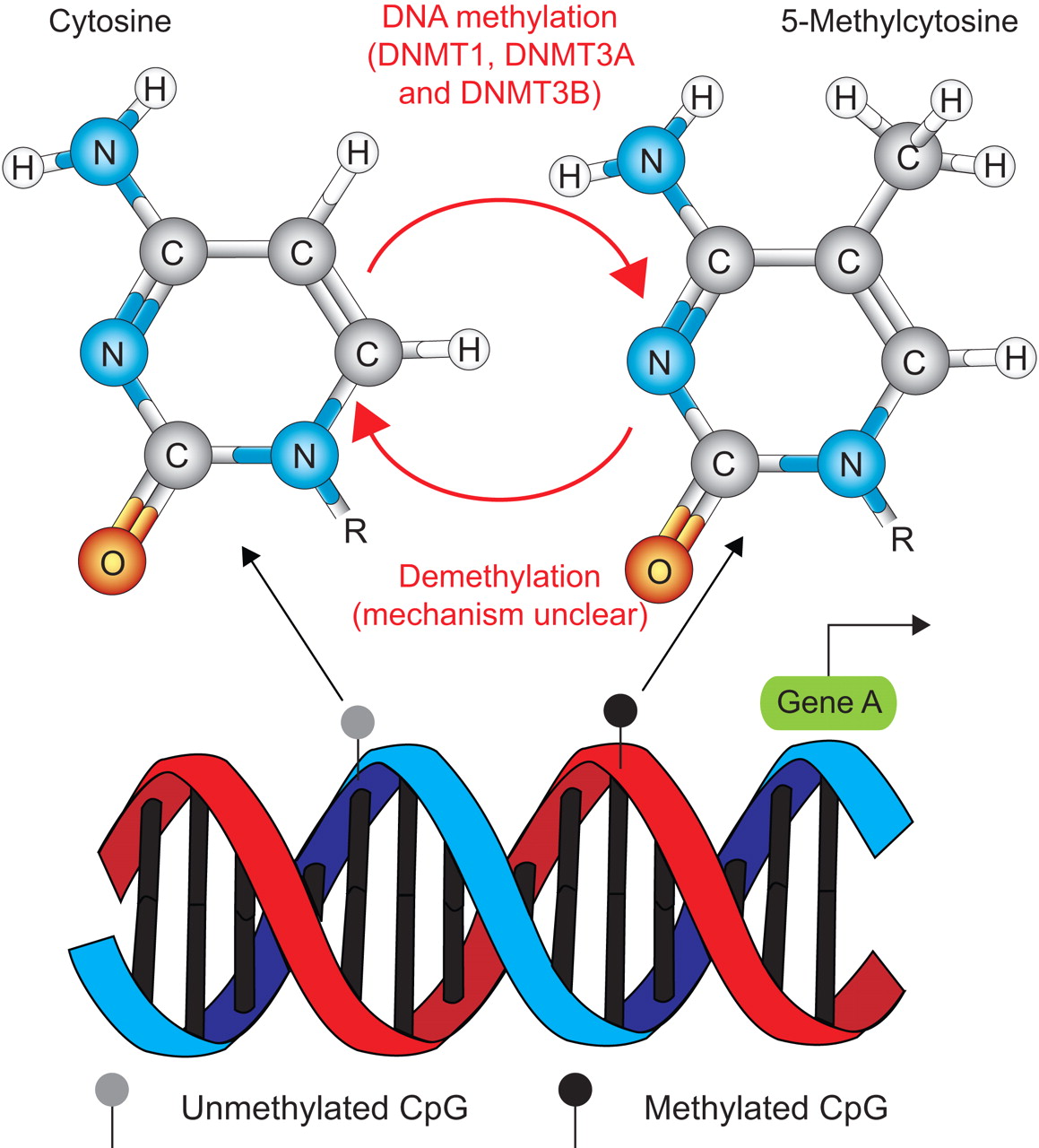
The principle of methylation and the chemical structure of cytosine and 5-methylcytosine. R is the connection to the DNA backbone.
This regulation is controlled in a tissue- and developmental stage-specific manner. Thus, mechanisms of epigenetic regulation and the degree of epigenetic modification vary between different cells and tissues (e.g. between lung epithelial cells and blood). Sufficient and balanced quantities of several diet-ingested molecules, such as folic acid, vitamin B12, choline, betaine and niacine, are important for the correct establishment and maintenance of epigenetic modifications throughout the genome. DNA methylation levels correlate with the levels of available folate and the genotype-dependent activity of enzymes involved in methylation, such as the 5,10-methylenetetrahydrofolate reductase gene 6. So far, four DNA methyltransferases have been identified (DNMT1, DNMT3A, DNMT3B and DNMT2) as well as a DNMT-related protein (DNMT3L) 7, 8. The first three catalyse the transfer of a methyl group from the universal methyl donor S-adenosyl-l-methionine to the cytosine base in DNA. Put simply, DNMT1 acts as maintenance methyltransferase as it has a preference for hemi-methylated templates. It is located at the replication fork during the S phase of the cell cycle and methylates the newly synthesised DNA strand using the parent strand as a template. Consequently, it passes on epigenetic information over cell generations. De novo methylation is carried out by the methyltransferases DNMT3A and DNMT3B. These enzymes have a preference for specific targets but also work cooperatively to methylate the genome. Possible trigger mechanisms to initiate de novo methylation include preferred target DNA sequences, RNA interference, certain chromatin structures induced by histone modifications and other protein-protein interactions 7, 8.
Although active demethylation of DNA undoubtedly occurs during development, the exact mechanisms for global and gene-specific demethylation events are still unclear and subject to much debate; all postulated mechanisms are not very efficient, indicating that the underlying mechanisms might be more complex than currently understood 9, 10.
Regulation of transcription by epigenetic modifications
Transcription does not occur on naked DNA but in the context of chromatin, which critically influences the accessibility of the DNA to transcription factors and the DNA polymerase complexes. DNA methylation, histone modifications and chromatin remodelling are closely interwoven and constitute multiple layers of epigenetic modifications for the control and modulation of gene expression through chromatin structure 11. DNMTs and histone deacetylases (HDACs) are found in the same multi-protein complexes and methyl-binding proteins interact with HDACs and histone methyltransferases, as well as with the chromatin remodelling complexes. In many cases DNA methylation follows changes in the chromatin structure and is used as the molecular mechanism to permanently and, thus, heritably lock the gene in its inactive state 4.
Cytosine methylation of CpG dinucleotides is found in close proximity to critically important cis-elements within promoters and is often associated with a repressed chromatin state and inhibition of transcription. In many cases, methylated and silenced genes can be reactivated using DNA methylation inhibitors such as 5-azacytidine. Methylation can interfere with transcription by inhibiting the binding of transcriptional activators with their cognate DNA recognition sequence such as Sp1 and Myc through steric hindrance. In addition, methylcytosine binding proteins (MBDs, MeCP2, UHRF, Kaiso and ZBTs) recruit transcriptional corepressors, such as histone deacetylating complexes, polycomb proteins and chromatin remodelling complexes, and attract chromodomain binding proteins to establish a repressive chromatin configuration 12.
Relevance of DNA methylation
Cytosine methylation is essential for mammalian embryogenesis, during which methylation levels change dynamically 13, 14. Epigenetic modifications are of particular importance for imprinted genes, i.e. a subset of genes that is asymmetrically expressed from only the maternal or the paternal allele in a parent-of-origin-specific manner in all somatic cells of the offspring 15. These imprinted genes are generally located in clusters and the alleles are differentially marked by DNA methylation, histone acetylation/deacetylation and histone methylation and often associated with antisense RNAs 16, 17. Imprinted genes are probably the most important buffering factors for regulating the day-to-day flux between mother and fetus in placental mammals. DNA methylation plays an important role in the maintenance of genome integrity by transcriptional silencing of repetitive DNA sequences and endogenous transposons 18. Random silencing of one of the two X chromosomes in embryonic tissues of female mammals to achieve dosage compensation is another paradigm for a stable and heritable epigenetic state in somatic cells 19.
Epigenetics holds promise for the explanation of at least a part of the influences the environment has on a phenotype. Studies in monozygotic twins demonstrated that epigenetic differences in such genetically identical humans accumulate with age and different environments create different patterns of epigenetic modifications 20. Differences are, therefore, largest in twin pairs of old age that have been raised separately. Transient nutritional or chemical stimuli occurring at specific ontogenic stages may have long-lasting influences on gene expression by interacting with epigenetic mechanisms and altering chromatin compaction and transcription factor accessibility. In particular, modifications to the environment during early development can lead to permanent changes in the patterns of epigenetic modifications. Epigenetics might, therefore, provide a mechanism by which physiological homeostasis could be developmentally programmed and inherited.
EPIGENETICS AND DISEASE
DNA methylation and chromatin structure have been found to be strikingly altered in many pathological situations, particularly cancer and various mental retardation syndromes. Altered levels of folate and homocysteine have been repeatedly linked to disease. Even so called monogenetic diseases, such as α-thalassaemia, that have previously been attributed solely to genetic alterations, can be caused by epigenetic alterations at the same locus 21.
Cancer is probably the most extensively studied disease with a strong epigenetic component 22. In tumours a global loss of DNA methylation (hypomethylation) of the genome is observed. The overall decrease in DNA methylation is accompanied by a region- and gene-specific increase of methylation (hypermethylation) of multiple CpG islands, often associated with transcriptional silencing of the associated gene. Genes of numerous pathways involved in signal transduction, DNA repair, detoxification, cell cycle regulation, differentiation, angiogenesis and apoptosis are often inappropriately inactivated by DNA methylation. Methylation analysis can be carried out in cancer tissue itself but, more importantly, recent reports have demonstrated a high level of concordance of DNA methylation patterns in tumour biopsies and matched DNA samples extracted from body fluids such as serum, plasma, urine and sputum 23, 24. As DNA methylation is a non-mutational and, therefore, at least in principle, a reversible modification, it can be used as point of departure for anti-neoplastic treatment by chemically or antisense oligonucleotide-induced demethylation 25.
DNA METHYLATION AND COMPLEX DISEASE
While most interest has been focused on epigenetic mechanisms in cancer, it is very likely that epigenetic changes also contribute directly or indirectly to the development of many complex and multifactorial diseases. Genetic variations so far identified to confer susceptibility to complex diseases only explain a small fraction of the disease risk. Epigenetic mechanisms are capable of explaining various non-Mendelian features of multifactorial diseases, such as the relatively high degree of discordance in monozygotic twins. Undoubtedly, environmental factors also play a critical role in triggering the onset of complex diseases and epigenetic modifications constitute a memory of the stimuli or insults an organism has been exposed to. However, only a few of these complex diseases have so far been studied in detail with regard to their epigenetic component 26.
Epigenetic modifications might mediate adaptation of the organism to its environment, starting early in life 27. Nutritional stress during early development seems to trigger systemic epigenetic modifications in mammals, especially of the metabolic gene network 28. The modulation of epigenetic patterns in utero has given rise to the hypothesis that the in utero environment can cause permanent changes to metabolic processes, directly affecting postnatal phenotype and conferring susceptibility to multifactorial disease later in life 29, 30. There is also evidence for transgenerational inheritance of epigenetic patterns leading to complex diseases, as a similar effect was observed in individuals thriving in affluent conditions but whose parents or even grandparents were nutritionally deprived 31.
EPIGENETICS AND INFLAMMATORY/IMMUNE DISORDERS
Epigenetic changes are also found in chronic inflammatory diseases in which epigenetic changes induced by inflammation contribute to the pathogenesis of the disease; for example, atherosclerosis is characterised by an increased degree of hypomethylation in vascular tissue and blood cells correlating with the severity of the disease 32. Local DNA methylation alterations (hyper- and hypomethylation) have been found to precede aortic lesions in human cell line and mouse models, and might thus present a first stage of pathogenesis 33. DNA hypomethylation is also observed in synovial fibroblasts 34 and in T-cells 35 from patients with rheumatoid arthritis. Autoimmune diseases, such as systemic lupus erythematosus, including drug-induced lupus, are characterised by hypomethylation of certain T-cell populations but without concomitant genomic instability 36, 37.
EPIGENETICS, ASTHMA AND ALLERGY
The investigation of epigenetic changes involved in the pathogenesis of asthma and allergy is still at a very early stage. Epigenetics may explain how genetic susceptibility and exposure to environmental factors interact in defining atopic disease phenotypes. Methylation is the most extensively studied epigenetic mechanism in asthma- and allergy-related genes and environmental effects. Apart from methylation, altered histone modifications might also play a role in asthma. There is evidence for an increased activity of histone acetyltransferases (HATs) and a simultaneous reduction of HDAC activity in asthmatic patients 38, 39. Both of these histone modification mechanisms can lead to hyperacetylation of the N-terminal histone tails, opening up chromatin structure and increasing gene transcription by facilitating the binding of transcription factors 40. These modifications might potentially contribute to the increased expression of genes involved in inflammatory responses characteristic for asthma. Interestingly, hyperacetylation mechanisms were suppressed or even reversed when the asthmatics were regularly treated with inhaled steroids in some of those studies 39. These effects are not only observed in asthma but have also been well defined in chronic obstructive pulmonary disease (COPD). There, total HDAC activity was found to be related to the severity of the disease 41. One of the most prominent aspects in the development of asthma and allergy, in which epigenetic mechanisms are known to play an important role, is T-cell differentiation and regulation 42, 43, a crucial event in the onset of atopic disease.
EPIGENETICS OF T-CELL DIFFERENTIATION, A HALLMARK OF ALLERGY
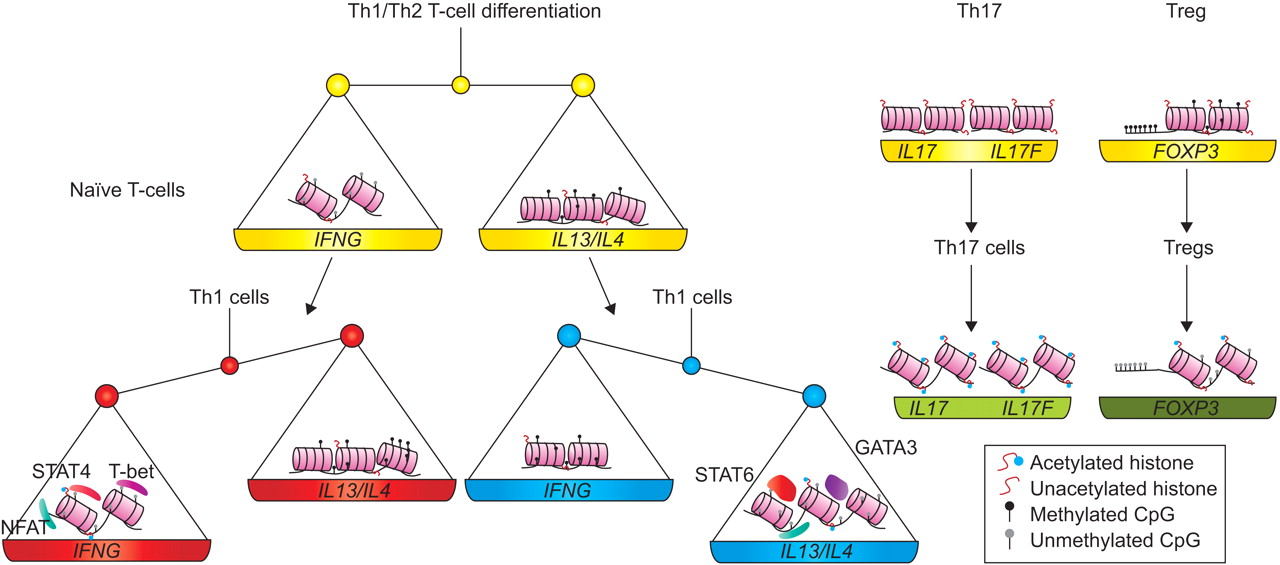
The development of a naïve T-cell into a T-helper type 1 (Th1) or 2 (Th2) cell is associated with lineage-specific changes in epigenetic patterns. During differentiation towards Th1 cells, de novo methylation of the intergenic region between interleukin (IL)4 and IL13 on chromosome 5q21 is observed 44, while histone acetylation remains at a low level comparable to that of naïve T-cells 45. Both effects lead to a reduced activity of Th2-specific genes in Th1 cells. Additionally, the binding of T-bet to the promoter of interferon gamma (IFNG) not only induces promoter activity itself but dislocates HDAC-containing complexes, which in turn enables HATs to bind 46. Likewise, the activity of the Th1-specific transcription factor (IFNG) is suppressed in Th2 cells by increased methylation of the IFNG gene locus, preventing nuclear factor of activated T-cells binding to the IFNG promoter 47. In contrast, the IL13/IL4 region undergoes extensive demethylation in Th2 lineage differentiation, which facilitates binding of the Th2-associated transcription factors STAT6 and GATA3 to the locus increasing expression levels of the respective genes 44. Simultaneously with the demethylation of the DNA, histones in the IL13/IL4 region become acetylated 45 and the higher-order chromatin structure of the locus changes (figs 3 and 4) 48.

Different patterns of epigenetic mechanisms influence the differentiation and the maintenance of T-cells into T-helper type 1 (Th1), 2 (Th2) and 17 (Th17) cells or regulatory T-cells (Tregs), respectively.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
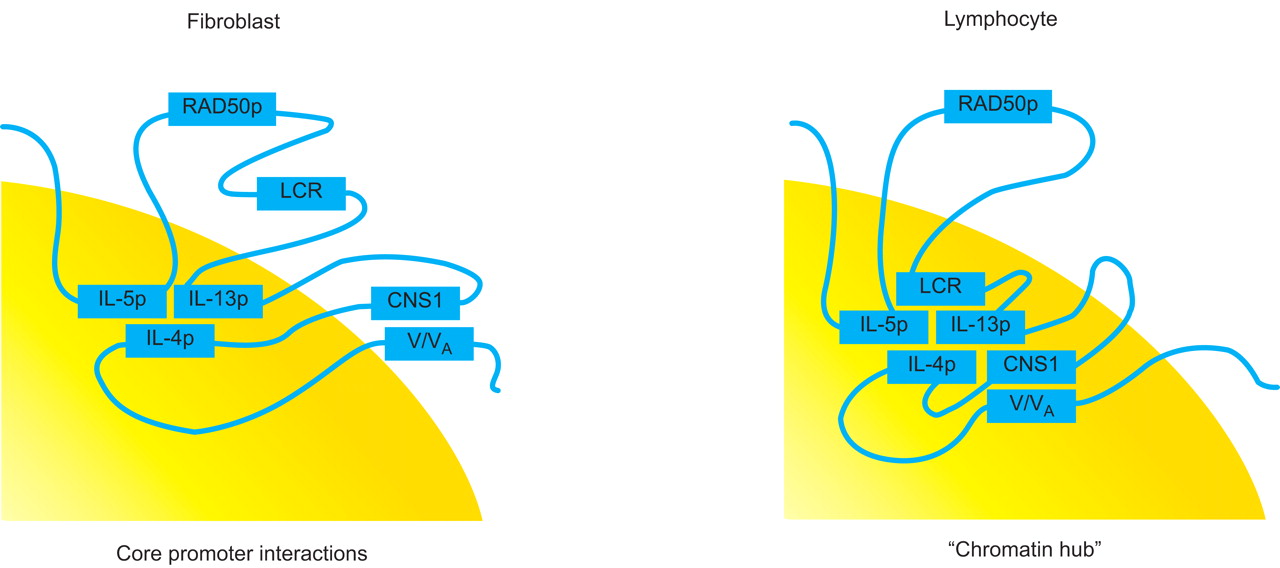
Complex three-dimensional structure of the interleukin (IL)13–IL4 locus. The cytokine gene promoters of IL5, IL13 and IL4 are suggested to interact with each other. This interaction is always present even if these cytokines are not transcribed, like in fibroblasts. For a transcription an interaction with additional intergenic regions in a “chromatin hub” is necessary as shown here in lymphocytes. Reproduced from 48 with permission from the publisher.
Regulatory T-cell (Treg) development and function is predominantly mediated by FOXP3, a member of the forkhead/winged-helix family of transcription factors. Several studies have shown that at least in mice FOXP3 is influenced by various epigenetic mechanisms. An enhancer upstream of FOXP3 is methylated in naïve CD4+CD25- T-cells, activated CD4+ T-cells and transforming growth factor β-induced adaptive Tregs, but completely demethylated in natural Tregs 49. Furthermore, the demethylation of an evolutionary conserved noncoding region upstream of exon 1 is necessary for stable FOXP3 expression 50. As HDAC inhibitors enhance FOXP3 expression, it was also suggested that the level of histone acetylation of the FOXP3 locus plays a role in Treg development 51. Histone acetylation may be involved in T-cell differentiation into Th17 cells as well. In Th1 and Th2 cells, the promoter regions of IL17 and IL17F were hypoacetylated, favouring the absent expression of these genes. However, in Th17 cells producing considerable amounts of IL17 and IL17F, increased histone acetylation has been detected 52.
Epigenetic mechanisms may also play a role in the regulation of cell-specific gene expression patterns of allergy-associated cytokines beyond T-cells. In mast cells, the activation of the IL4/IL13 locus is also regulated via epigenetic modulation (e.g. reviewed by Ansel et al. 53). However, epigenetic mechanisms in that region vary between mast cells and T-cells. While for T-cells some knowledge on the involvement of epigenetic modifications in cell commitment and function has been acquired, little is yet known for other cell types involved in asthma and allergy.
ENVIRONMENTAL FACTORS POTENTIALLY AFFECTING EPIGENETIC MECHANISMS IN ASTHMA
In addition to genetic susceptibility, environmental factors play an important role in the pathogenesis of asthma and allergy. Gene by environment interactions are thought to occur and to significantly influence disease development 54. Epigenetic factors may mediate at least some of these environmental effects 55. Thus, we systematically examined whether environmental factors known to have an impact on asthma and allergy have also been reported to induce epigenetic changes in experimental settings (table 1).
Smoke exposure and epigenetics
The single most consistent and significant environmental risk factor for the development of childhood onset asthma identified in numerous epidemiological studies is the exposure to tobacco smoke. Recent findings demonstrated that direct inhalation of cigarette smoke alters DNA methylation patterns in lung derived cells of mice shortly after exposure 63. In humans, cancer-related studies 67, 68 reported gene promoter hypermethylation in sputum of cancer-free heavy smokers, suggesting the presence of epigenetic field defects prior to pathologically detectable cancers. Several studies associate in utero tobacco smoke exposure, smoking of the mother previous to pregnancy and even smoking by the grandmother to occurrence of asthma in the child or the grandchild, respectively 57, 69, 70. Parental smoking affects the methylation of CpGs in the vicinity of numerous genes investigated in DNA collected from buccal cells of children 64. Methylation was increased for AluYb8 and LINE1 repetitive elements and decreased for the promoter regions of AXL and PTPRO genes. Interestingly, genetically determined deficiency and genetic variation in detoxification genes of the glutathione S-transferase (GST) family seem to modify smoke-associated methylation effects 71. This may be explained by the fact that GST genes detoxify numerous cancerogenic compounds, which can facilitate methylation. Thus, genomic variations in GST genes could determine the level of epigenetically active substances acting on CpGs in disease-associated candidate genes. Furthermore, it has been shown that transcriptional activity of GST promoters depends on genetic variants and leads to differential DNA methylation susceptibility of the respective promoter, as recently demonstrated in a study of locally advanced breast cancer 72.
Ozone and oxidative stress
Exposure to elevated ozone levels is a well known trigger for exacerbation in asthma patients. The role of ozone in disease development is controversial. The hypothesis and the evidence that there may be a causal relationship between oxidative stress, DNA methylation and carcinogenesis was recently reviewed by Franco et al. 73. A correlation between DNA methylation and oxidative stress has been proposed for Alzheimer disease 74. Whether ozone-related epigenetic changes are involved in the development of asthma is still unknown.
Traffic exposure
An impact of traffic exposure on asthma development via epigenetic mechanisms was described by Perera et al. 75. They studied the influence of exposure to traffic-related air pollution in mothers on asthma development in their children. A correlation between a high exposure to polycyclic aromatic hydrocarbons (PAH) of the mothers and asthma was reported, which seemed to involve the DNA methylation of the ACSL3 promoter. There, a higher PAH exposure leads to higher levels of methylation. A less active ACSL3 may diminish fatty acid utilization and beta-oxidation energy production, and may also have an impact on the phospholipid composition of cell membranes.
Endotoxin and farm exposure
Endotoxins are bacterial wall components, highly immunogenic and abundant in the environment. They are thought to be a good proxy for microbial exposure. In farm studies, protection against hay fever and atopic wheeze has been associated with high exposure levels of endotoxins 76. Their role in asthma and allergy development is controversial and a causal relationship between endotoxin exposure and protection against allergy and asthma has not yet been established. Nonetheless, there are indications for gene by environment interactions, where differences in endotoxin levels affect associations between CD14 promoter polymorphisms and immunoglobulin (Ig)E mediated allergy 61, 62. Endotoxin levels and farm exposure also influence the effect of toll-like receptor (TLR) polymorphisms on asthma and allergy in rural populations 59. Furthermore, farm exposure (even prenatally) seems to influence the expression of innate immunity-related receptors and molecules such as TLR2, TLR4 and CD14 60, 76. Even though actual data are still missing, it would not be surprising if epigenetic mechanisms contribute substantially to these environmentally induced changes in gene expression. A single study investigating epigenetic effects of endotoxins on tumour necrosis factor (TNF) expression suggests that indeed endotoxins modulate TNF gene silencing by specific chromatin remodelling 66. In addition it could be shown that histones H3 at the IL-1β promoter undergo demethylation after endotoxin stimulation in normal human leukocytes 77. As these alterations were not observed in endotoxin-tolerant cells, this may indicate that epigenetic mechanisms might be involved in innate immunity response triggered by endotoxins.
Diet
An association between asthma-related phenotypes, DNA methylation and diet was proposed by Hollingsworth et al. 78. The effect of a diet rich in methyl donors (folic acid) was studied in mice. The offspring of mice treated with folic acid displayed higher levels of airway hyperactivity, increased airway eosinophilic inflammation and higher levels of IgE production. The methyl-rich diet decreased transcriptional activity and mRNA expression of several genes, which was accompanied by DNA hypermethylation of the respective promoters. One of these differentially methylated genes was RUNX3, a gene linked to silencing of IL4 and activation of FOXP3 79. It could thus be speculated that changes of RUNX3 activity could influence allergic airway diseases.
The effects of a diet rich in folate on developing asthma was studied recently in an Australian prospective birth cohort. There, the intake of supplemental folic acid during the last months of pregnancy increased the risk of the child developing asthma 80. Based on prior experimental data of folic acid in epigenetic studies, the authors speculated that the epidemiological effects observed may be epigenetic. However, no proof for this hypothesis was provided.
Folate is not the only dietary supplement suspected to display epigenetic effects. High calcium intake has been reported to influence associations of genetic variants with breast cancer in the gene coding for the vitamin D receptor (VDR) 81. In choriocarcinoma cells the activity of VDR was found to be probably epigenetically controlled, as silenced VDR could be activated through the inhibition of either DNA methylation or histone deacetylation 82. These observations suggest that VDR is involved in epigenetic mechanisms, an interesting observation as genetic polymorphisms in VDR have been associated with asthma and airway hyperresponsiveness 83. Reduced levels of vitamin D have been shown to be associated with elevated markers of allergy and asthma severity 84.
However, not only the intake of dietary supplements but also malnutrition or undernourishment influences epigenetic regulation and subsequent development of disease. In a cross-sectional study of African-Americans high fat consumption and diabetes was associated with a hypermethylated RARβ2 promoter 85. In undernourished rats, a relationship between the amount of protein in the nutrition and the methylation of the AT1b angiotensin II receptor gene in their offspring 86 was observed. The progeny of the study group fed with substantially decreased amounts of protein showed a demethylation of the AT1b promoter. Demethylation of the promoter led to elevated expression of AT1b, which was suggested to be causal for the hypertension observed in these rats.
In humans, famine in previous generations may affect methylation in offspring as it was suggested in a study investigating potential DNA methylation alterations in Dutch individuals whose parents were exposed to a period of severe famine at the end of World War II. There, changes in methylation of IL10, an important regulator of inflammation potentially involved in asthma and allergy development, were observed 87. Children concerned by the famine showed, in contrast to their unexposed siblings, a hypermethylated IL10 promoter.
PUTATIVE TARGET GENES FOR EPIGENETIC MODIFICATION IN ASTHMA
After analysing published evidence for epigenetic effects on asthma induced by environmental factors, we also investigated putative asthma candidate genes for their potential to be regulated by epigenetic mechanisms. Three strategies were applied to systematically identify: 1) genes harbouring single nucleotide polymorhisms that showed associations with asthma in at least five independent studies based on extensive in silico data mining; 2) genes that had been suggested by a recent comprehensive review of the field to be one of 10 important asthma candidate genes 88; and 3) asthma genes recently identified by genome-wide association studies (GWAS) 89–92, even though only very limited data are yet available for GWAS genes.
Asthma candidate genes selected according to these three aforementioned criteria for which evidence for epigenetic regulation exists are summarised in table 2. Putative asthma candidate genes for which no published evidence for epigenetic regulation was found are ADRB2, CD14, CHI3L1, CHRNA2, DENND1B, GSDMA, HLA-DRB1, IL1RL1, IL33, MYB, NPSR1, PSMD3, TLE4 and WDR36.
The epigenetic regulation of the IL4/IL13 locus on chromosome 5q31 has been extensively studied. As these genes have been studied in the context of T-cell regulation and differentiation, these studies have already been discussed in a previous section of this review. In addition, Kwon et al. 96 claimed that CpG methylation in the IL4 and IFNG promoters differed in asthmatics and non-asthmatics after stimulation with phytohaemagglutinin. However, their observation was limited to three cases and three controls only. Obviously, additional studies for validation are required before conclusions can be drawn on whether methylation patterns in this locus differ between asthmatics and nonasthmatics.
For ADAM33 it could be shown that the promoter region of the gene is differentially methylated in epithelial cells and fibroblasts 93. However, these differences were independent of asthma status in the analysed samples. Methylation patterns of CpGs related to MS4A2, coding for the beta-chain of the IgE high-affinity receptor, were recently investigated in people with and without atopy 98. Variation in methylation at this locus in peripheral blood lymphocytes was not associated with age, sex, daily steroid use or atopic status, and there were no differences in methylation between mothers and fathers of atopic cases. Both these studies stress the importance of selecting specific cell subsets for epigenetic analysis that are relevant for disease development. As it is not yet clear which cells are crucial for asthma and atopy development (epithelial cells versus immune cells; cells in circulation versus those at the locus of inflammation), epigenetic analyses in asthma and allergy are severely limited.
For a region on chromosome 17q21 strongly associated with childhood asthma in the first GWAS for asthma (and numerous subsequent replication studies) 91, some evidence for epigenetic regulation exists. The locus contains ORMDL3, GSDMB and ZPBP as well as other genes. In a recent study, genetic variants associated with asthma affected chromatin remodelling of a regulatory element controlling accessibility to the entire locus 102. Furthermore, the expression of IKZF3, another putative candidate gene in the 17q21 region coding for a zinc finger with potential immunogenic effects, may also be influenced by epigenetic mechanisms. Evidence derives from tumour cell studies, in which IKZF3 displays differential methylation in cell lines and its expression is silenced by methylation of the CpG island associated with the transcription start site 94.
For PDE4D, a further asthma candidate gene suggested by a GWAS 90, epigenetic data had been acquired previously to the GWAS. PDE4D encodes several isoforms and the expression of PDE4D1 and PDE4D2 is regulated by an intronic PDE4D promoter. At least in vascular smooth muscle cells, experiments based on chromatin immunoprecipitation showed phenotype-specific gene expression influenced by histone acetylation 100. The promoter was acetylated only in vascular smooth muscle cells derived from damaged blood vessels, whereas in cells derived from healthy arteries, histones were not acetylated. No data exists on immune cells or lung specific effects.
ALTERATIONS OF miRNA EXPRESSION IN ASTHMA
miRNAs are small single-stranded noncoding RNAs that are highly conserved across species and function as endogenous regulators of numerous target genes 103. Hundreds of human miRNAs localised in the introns of protein-coding genes or in the noncoding regions of the genome have been identified in the human genome. They are expressed in a tissue-specific manner and play important roles in cell proliferation, apoptosis, and differentiation during mammalian development. miRNAs are able to negatively regulate expression through degradation of mRNA transcripts or inhibition of protein translation. It has been estimated that up to 30% of all genes might be regulated through miRNAs 104. miRNAs play also an important role in the regulation of the immune system and the differentiation of the different lineages of the haematopoietic system 105. Aberrant expression of miRNAs may contribute to the development and progression of many human diseases, including asthma. For example, miR-223 might be involved in granulocyte production and the inflammatory response, as a mouse model mutated for miR-223 displayed neutrophilic inflammatory lung pathology and increased tissue destruction after endotoxin challenge 106. In another mouse model of house dust mite-induced allergic asthma, the asthmatic inflammation was found to be associated with a characteristic miRNA expression profile, in particular upregulation of miR-126 which suppressed the effector function of lung Th2 cells 107. Whether the observed changes in the mouse models are relevant to human asthma has so far not been investigated. A study analysing 227 miRNAs in airway biopsies from normal and mild asthmatic patients revealed cell type-specific miRNA profiles but no miRNAs associated to asthma 108.
CRITICAL ASSESSMENT AND OUTLOOK
Epigenetic studies in asthma are, as in many other complex diseases, at a very early stage. It would be surprising if epigenetic regulation would not be involved in the development and the natural course of a disease like asthma which is driven by environmental as well as genetic susceptibility factors. However existing epigenetic data is sparse (so far). Most of those studies were focused on mechanisms related to asthma (like T-cell lineage commitment) rather than on disease development itself. Reasons for that are manifold.
Asthma is a syndrome rather than a homogenous disease, involving numerous mechanisms acting in concert with each other, in parallel to each other, or against each other. Many different clinical phenotypes are summarised under the umbrella of an asthma diagnosis. While these asthma phenotypes share common clinical features, their molecular pathogenesis may differ greatly. This is of lesser concern when strong effects are studied in very large epidemiological or genetic studies on thousands of individuals. There, strong effects from small subpopulations may become significant enough to be identified. In epigenetics as it is studied today, numerous conditions have to be optimised a priori to detect epigenetic effects. In genetic studies, it does not matter from which cell type or cell state genomic DNA derives. The genetic code is universal and uniform in almost all cells. Epigenetic regulation has a different function. It allows for flexibility in the interpretation of DNA based information in specific cells and serves as a “flash memory” by transforming environmental effects into epigenetic patterns. This is the very reason why studying epigenetics is of interest in complex diseases. At the same time it causes serious challenges to epigenetic studies.
Choosing the right tissue in which to study epigenetics in complex diseases is a difficult task. Because cell types differ in their epigenetic patterns, an undefined mixture of cells is unarguably difficult to analyse. Thus, whole blood samples, while easy to obtain in large numbers in epidemiological settings, have to be reviewed critically in studies of an airway disease such as asthma. Lung tissue or well-defined cell types closely correlated to the pathogenesis of asthma would be ideal. However, so far it remains unclear which cells, cell types or tissues are involved in the onset of asthma.
Therefore, different (parallel) strategies to study asthma epigenetics are conceivable for the future. One possibility would be to vigorously dissect asthma phenotypes and to investigate epigenetic patterns in all variable sources of tissue in these highly selected individuals. While scientifically satisfying the drawback of this approach is the enormous efforts and costs needed to achieve sample collections of a size necessary for epigenetic studies in asthma subpopulations. Also, it may prove impossible to acquire relevant numbers of lung tissue samples in children. An alternative study design is to investigate effects of strong environmental factors identified to influence asthma development, such as microbial, allergen or smoke exposure, on epigenetics. With this approach, sample collection of considerable size could be established rather easily. These would be sufficient to study epigenetics on a broad basis in numbers large enough to level out false-positive associations and to identify subgroup effects. How feasible whole blood samples would be in such approaches has to be seen. The question remaining for both designs is what epigenetic effects to study, and in which genomic regions.
Also, the complexity of epigenetics is just starting to unravel. Methylation is not the only mechanism involved in epigenetic regulation. However, it is the one best understood and most accessible to high throughput analyses. Focusing on analysing DNA methylation patterns is a reasonable first approach. However, it may turn out not to be the epigenetic modification most important in asthma related mechanisms. Pyrosequencing bisulphite treated DNA is state of the art today, capable of handling relatively large sample numbers, but also this technique is limited to sequences of ∼300 base pairs in length 109. Thus, pyrosequencing is more feasible for validation than for scanning the epigenome for novel associations.
At this point, epigenetic studies in complex diseases rely heavily on experience gathered in the cancer field, where epigenetic mechanisms have already been investigated for a long time. A large number of genome-wide methods for DNA methylation analysis have been developed and due to the advent of novel sequencing technologies they become ever more powerful 110. CpG islands and transcription start sites may be important in cancer but could prove to be less important sites of epigenetic regulation in complex diseases 111. Thus, many of the currently available techniques might not yet have the required quantitative and spatial resolution to detect the subtle changes that might be implicated in complex diseases such as asthma. To our knowledge, no genome-wide DNA methylation study has so far been performed and published in asthma. Genome-wide bisulphite sequencing would constitute the ideal method for the unbiased detection of alterations in DNA methylation patterns 112, but the increased genome size (the genome size doubles as the two DNA strands are no longer complementary) in combination with the reduced sequence complexity and the required sequencing depth to achieve sufficient quantitative resolution makes it currently a daunting and cost-prohibitive endeavour for the analysis of several epigenomes. The rapid technological evolution, the expected decline in sequencing costs and the potential for direct read-out of methylated cytosines by some technologies under development (e.g. nanopores) will probably make these studies feasible in the next few years. Thus, it is important for the field to move on and to establish novel techniques and approaches to study epigenetics in complex diseases specifically. It will remain a challenge for the next few years.
Acknowledgments
This systematic review was supported by funding from the European Union, EU FP7 KBBE-2007-2-2-06 (EFRAIM). S. Michel is also supported by a PINA fellowship. Chemical structures were drawn with Jmol (an open-source Java viewer for chemical structures in 3D; www.jmol.org).
Footnotes
Previous articles in this series: No. 1: Hedlin G, Bush A, Lødrup Carlsen K, et al. Problematic severe asthma in children, not one problem but many: a GA2LEN initiative. Eur Respir J 2010; 36: 196–201. No. 2: Xepapadaki P, Papadopoulos NG. Childhood asthma and infection: virus-induced exacerbations as determinants and modifiers. Eur Respir J 2010; 36: 438–445. No. 3: de Groot EP, Duiverman EJ, Brand PLP. Comorbidities of asthma during childhood: possibly important, yet poorly studied. Eur Respir J 2010; 36: 000–000.
Statement of Interest
A statement of interest for M. Kabesch can be found at www.erj.ersjournals.com/misc/statements.dtl
- Received February 4, 2010.
- Accepted April 20, 2010.
- ©2010 ERS
REFERENCES











Jump To
- Article
- Abstract
- DNA METHYLATION
- EPIGENETICS AND DISEASE
- DNA METHYLATION AND COMPLEX DISEASE
- EPIGENETICS AND INFLAMMATORY/IMMUNE DISORDERS
- EPIGENETICS, ASTHMA AND ALLERGY
- EPIGENETICS OF T-CELL DIFFERENTIATION, A HALLMARK OF ALLERGY
- ENVIRONMENTAL FACTORS POTENTIALLY AFFECTING EPIGENETIC MECHANISMS IN ASTHMA
- PUTATIVE TARGET GENES FOR EPIGENETIC MODIFICATION IN ASTHMA
- ALTERATIONS OF miRNA EXPRESSION IN ASTHMA
- CRITICAL ASSESSMENT AND OUTLOOK
- Acknowledgments
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics