



Asthma and pulmonary arterial hypertension: do they share a key mechanism of pathogenesis?
- 1Pulmonary and Critical Care Medicine, Stony Brook University, Stony Brook, NY, 2Dept of Veterans Affairs Medical Center, Northport, NY, and 3Dept of Cell Biology and Physiology, University of New Mexico, Albuquerque, NM, USA.
- S. I. Said, Pulmonary and Critical Care Medicine, Stony Brook University; T-17-040, Stony Brook, 11794 NY, USA. E-mail: sami.i.said{at}stonybrook.edu
Abstract
Although largely distinct and seemingly unrelated, asthma and pulmonary arterial hypertension (PAH) have important pathological features in common, including inflammation, smooth muscle contraction and remodelling.
We hypothesised that these common features could be explained by one shared mechanism of pathogenesis: activation of the transcription factor NFAT (nuclear factor of activated T-cells). If this concept is validated, it could lead to the introduction of novel therapeutic strategies against both lung disorders.
In several experimental models, airway remodelling is accompanied by remodelling of smaller pulmonary arteries, validating the hypothesis of their similar pathogenesis. In addition, lungs of vasoactive intestinal peptide (VIP) knockout mice express airway hyperresponsiveness with airway inflammation and PAH with vascular remodelling, with both sets of pathological findings being reversible with VIP treatment. Preliminary data suggest that absence of the VIP gene leads to activation of the calcineurin–NFAT pathway, and that VIP is probably a physiological inhibitor of this pathway.
Enough evidence exists to support the views that asthma and PAH share important pathological features, probably related to NFAT activation, and that VIP may be a physiological modulator of this mechanism.
- Asthma
- pulmonary hypertension
- remodelling
- transcription factor
Asthma, a disease of the airways, and pulmonary arterial hypertension (PAH), a disease of the pulmonary circulation, have markedly different clinical presentations. On closer examination, however, the two diseases can be shown to share several key pathological features.
Asthma is a multifaceted disease characterised by airway hyperresponsiveness (AHR), airway inflammation and varying degrees of smooth muscle cell proliferation termed airway remodelling 1–7. Research has often focused on one or another of the component elements of the asthmatic response, and corresponding therapeutic initiatives have been proposed with varying degrees of success. However, the disease continues to present challenges to physicians and investigators alike. The very complexity of the disease has stimulated the search for more comprehensive answers, and even prompted the question: can a single basic mechanism be at the root of the major features of asthma? Herein, we review evidence for the existence of such a mechanism, one that might also help explain the basic pathological processes of PAH.
COMMON PATHOLOGICAL FEATURES IN ASTHMA AND PAH
Both asthma and PAH exhibit three major pathological features: inflammation, smooth muscle constriction, and smooth muscle cell proliferation.
Inflammation has long been acknowledged as a cardinal feature of the asthmatic response 1, 7, 8, and has been gaining increasing recognition as a significant component of clinical and experimental PAH phenotypes 9. In both diseases there is increased resistance in, and narrowing of, airways and pulmonary arteries, respectively, due to airway and pulmonary vasoconstriction 10, smooth muscle constriction, and thickening of the walls caused by smooth muscle and other cell proliferation known as remodelling. Muscularisation and remodelling of smaller pulmonary arteries are essential pathological lesions in PAH 11, 12. Asthmatic airway inflammation causes tissue injury and related structural changes that may lead to airway remodelling, as described in humans and in experimental models in mice and rats 13–20. Remodelling includes an increase in airway wall thickness, fibrosis, smooth muscle mass and vascularity, as well as abnormalities in extracellular matrix composition 17, 21. Recent reviews have addressed the molecular mechanisms, consequences and significance of airway remodelling, along with its lack of responsiveness to current pharmacological therapy 5–7, 22–25.
These shared pathological features between asthma and PAH are unlikely to be strictly coincidental. Rather, they probably reflect a single underlying mechanism that is common to both diseases. Compelling evidence points to one particular transcription factor as playing a key role in this mechanism.
CALCINEURIN–NUCLEAR FACTOR OF ACTIVATED T-CELLS SIGNALLING
Transcription factors of the NFAT (nuclear factor of activated T-cells) family are master regulators of immune responses, as well as of gene expression in a host of tissues and organs, including smooth muscle (fig. 1⇓). Four of the NFAT proteins (NFATc1, NFATc2, NFATc3 and NFATc4) are normally cytoplasmic, i.e. inactive. However, upon dephosphorylation by the Ca2+-responsive phosphatase calcineurin, the proteins are activated and translocated to the nucleus. Activated NFAT exerts its multiple regulatory actions by forming co-operative complexes with other transcription factors, including the activator protein-1 family of proteins 26.

The calcineurin–NFAT (nuclear factor of activated T-cells) pathway and its possible regulation by vasoactive intestinal peptide (VIP). In resting cells, NFAT resides in the cytoplasm, where it is maintained in a phosphorylated, inactive state. Stimulation of cell-surface receptors coupled to phospholipase C (PLC) results in calcium mobilisation, initially from intracellular stores, followed by influx of Ca2+ through specialised voltage-dependent Ca2+ channels (VDCC) and Ca2+ release-activated Ca2+ channels (CRAC). Upon activation by Ca2+, the phosphatase calcineurin dephosphorylates NFAT, triggering its activation and nuclear translocation. In the nucleus, NFAT interacts with other transcription factors, including activator protein (AP)-1, to stimulate gene expression. When NFAT is re-phosphorylated it is exported back to the cytoplasm. The immunosuppressive drug cyclosporin A inhibits calcineurin interactions with all its substrates, whereas VIVIT selectively inhibits NFAT activation. VIP appears to inhibit this pathway, but its mechanism of action has yet to be determined. IP3: inositol-1,4,5-triphosphate. Modified from 26.
THE ROLE OF NFAT ACTIVATION IN THE PATHOGENESIS OF INFLAMMATORY AND IMMUNOLOGICAL DISORDERS
NFAT proteins are expressed on T-cells and other classes of immune system cells, such as B-cells, mast cells, eosinophils and natural killer cells. NFAT proteins are activated by stimulation of Gq-coupled receptors coupled to Ca2+ mobilisation, such as antigen receptors on T- and B-cells, and receptors on other cells. NFAT transcription factors are key regulators of inducible gene transcription during the immune response. NFAT activation can also promote smooth muscle cell proliferation and, thus, contribute to the proliferative reaction in airway remodelling.
EVIDENCE LINKING NFAT ACTIVATION DIRECTLY TO ASTHMA PATHOGENESIS
In a model of allergic airway inflammation, transgenic mice expressing dominant-negative NFAT mutant exclusively in T-cells showed reduced antigen-specific T-helper (Th)2 antibody levels and interleukin (IL)-4 production by CD4+ T-cells. Furthermore, accumulation of eosinophils in bronchoalveolar lavage fluid was delayed and the mice were more resistant to the development of allergen-induced lung pathology 27. NFAT proteins play a central role in the transcriptional regulation of IL-13 in mast cells and Th2 cells, and IL-13 is both necessary and sufficient for experimental models of asthma 28. NFAT blockade by in vivo transfection of an NFAT decoy precluded Th2 cytokine expression and AHR in experimental mouse asthma 29. Pathophysiological features of asthma expressed in mice lacking the vasoactive intestinal peptide (VIP) gene are associated with, and attributable to, NFAT activation.
NFAT ACTIVATION IN THE PATHOGENESIS OF PAH
NFAT activation has been identified in experimental models of PAH, and in one clinical form of the disease, idiopathic PAH (IPAH). de Frutos et al. 30 presented evidence that NFATc3 activation mediates chronic hypoxia-induced pulmonary vascular hypertrophy and remodelling in mice, with α-actin upregulation. Koulmann, et al. 31 demonstrated the critical role of calcineurin activation in the production of hypoxia-induced PAH and right ventricular hypertrophy in rats. Both pathological findings were fully prevented by treatment with calcineurin inhibitor cyclosporin A. NFATc2 activation has been demonstrated in small- and medium-sized pulmonary arteries of rats with monocrotaline-induced PAH 32. Cyclosporin A reversed PAH in vivo and reversed several features of the PAH phenotype of pulmonary arterial smooth muscle cells (PASMCs), including decreased voltage gated K+ (KV) current and KV1.5 expression 32. Similarly, in IPAH patients, NFATc2 was activated in PASMCs, lung and T-lymphocytes. Inhibition of NFATc2 decreased PASMC proliferation and increased apoptosis in vitro 32. Selective inhibition of calcineurin–NFAT signalling in the lungs of VIP-/- mice attenuated the pathological changes of PAH (as well as of asthma) in these mice, and PASMCs showed predominant nuclear localisation, i.e. activation of NFATc (unpublished data).
COMBINED FEATURES OF ASTHMA AND PAH IN THE SAME EXPERIMENTAL MODELS
If the same molecular mechanism underlies the pathogenesis of both asthma and PAH, we might expect that, at least in some instances, both airway and vascular pathology might be expressed concomitantly in the same lung.
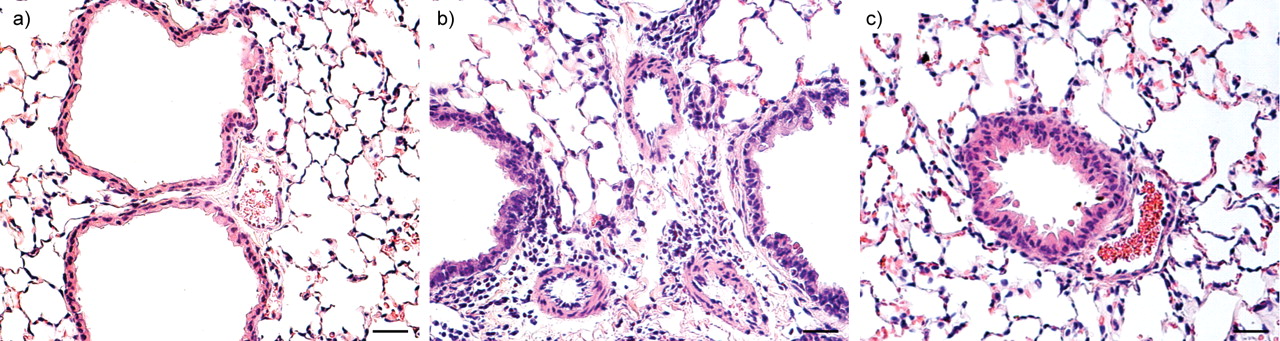
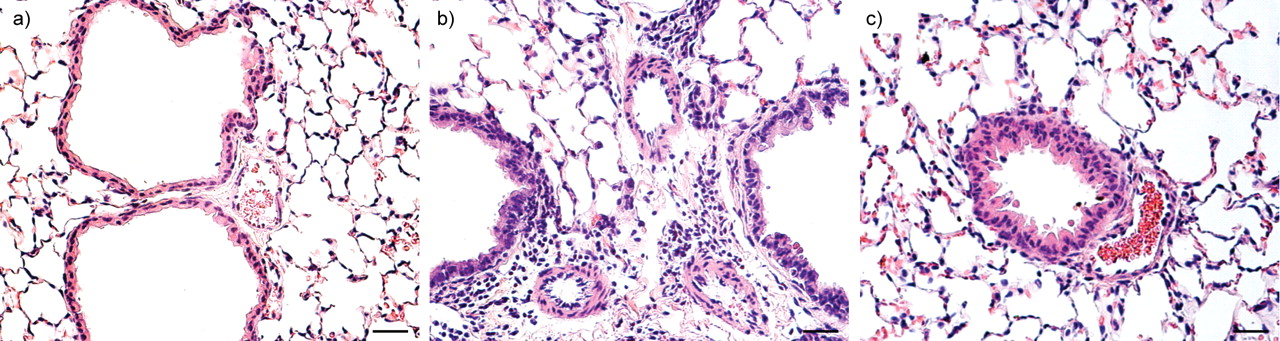
We first observed this combined pathology in mice lacking the VIP gene. As reported earlier, such mice simultaneously express AHR with airway inflammation, together with PAH, pulmonary vascular remodelling and perivascular inflammation (fig. 2⇓). Treatment of the mice with VIP reversed both sets of phenotypic changes, confirming that they result from the absence of the VIP gene 33, 34.
{kind=link}
{kind=link}
Haematoxylin and eosin lung sections from a) wild-type mice, b) vasoactive intestinal peptide (VIP)-/- mice and c) VIP-/- mice treated with VIP. Compared with lungs from controls, wild-type mice and lungs from VIP-/- mice showed peribronchiolar and perivascular lymphocytic and eosinophilic infiltrates, and marked medial thickening of smaller pulmonary arteries. After treatment with VIP (15 μg intraperitoneal every other day for 4 weeks for a total of 14 injections) both vascular thickening and inflammation were almost totally abolished. Scale bars = 25 μm.
In a model of Th2 immune response, C57BL/6 mice developed severe pulmonary arterial remodelling after immunisation and prolonged intermittent exposure to intranasal Aspergillus fumigatus 35. Despite the vascular remodelling there was no significant increase in pulmonary arterial pressure. This unexplained discrepancy raises the possibility that some asthmatics, such as these mice, might have unrecognised pulmonary vascular remodelling.
Female BALB/C mice that were sensitised and repeatedly challenged with ovalbumin developed remodelling of bronchi, as well as inflammation and remodelling of pulmonary vessels, both adjacent to 36 and some distance from allergen-exposed bronchi 37. There were notable similarities between the remodelling of airways and pulmonary vessels with regard to increases in smooth muscle mass, collagen synthesis and proliferation.
Vascular remodelling was also observed in the same strain of mice, following chronic exposure to house dust mite, a model that induces persistent airway inflammation consistent with that seen in human asthmatics 38.
As convincing as these observations are, they need to be shown to apply to human asthmatics as well as to mice 39. There have been only isolated reports of pulmonary vascular pathology in asthmatics. In a group of six subjects who died during an asthmatic attack, smaller pulmonary arteries showed “an important inflammatory process” in their walls, but normal medial and intimal thickness 40. Another study reported abnormalities in bronchial arteries in the lungs of asthmatics, but comparable changes in pulmonary arteries were not described 41.
DEVELOPMENTAL RELATIONSHIPS OF AIRWAY AND PULMONARY VASCULAR SMOOTH MUSCLE
The close relationship between the responses of airway and pulmonary vascular smooth muscle cells in the pathological settings of bronchial asthma and PAH, respectively, begins during embryological development of the lung. Thus, pulmonary vascular smooth muscle originates partly from migration of adjacent airway smooth muscle cells and from surrounding undifferentiated mesenchymal cells 42. As in remodelling, smooth muscle development and proliferation are controlled by the same or similar signalling molecules, including glycogen synthase kinase-3/β-catenin and the Wnt family 43–47. Although the signals that stimulate their formation may be distinct, airway and vascular smooth muscle cells are sufficiently alike to raise the question as to whether they can be differentiated from one another 48.
IS VIP A PHYSIOLOGICAL MODULATOR OF NFAT ACTIVATION?
In addition to highlighting shared pathological features and a probable mechanism of pathogenesis between bronchial asthma and PAH, the findings presented herein suggest an important physiological interaction between VIP and calcineurin–NFAT signalling. 1) Lack of the VIP gene results in two phenotypes attributable to NFAT activation: asthma and PAH (fig. 2⇑). 2) Replacement therapy with VIP reverses both phenotypes. 3) The therapeutic effect of VIP in VIP-/- mice duplicates that of the VIVIT peptide 49, a selective calcineurin/NFAT pathway inhibitor. 4) These findings suggest that lack of the VIP gene results in NFAT activation 50. 5) Preliminary studies indicate that VIP directly inhibits NFAT activation in transfected cells (unpublished data). 6) Taken together, these results strongly point to VIP as a negative regulator of calcineurin/NFAT signalling. 7) If confirmed, such a role for VIP could imply that it serves as a physiological defence against bronchial asthma and PAH. 8) As an anti-proliferative, anti-inflammatory agent, the peptide would also have a clear therapeutic potential in asthma and PAH, as well as in autoimmune disorders and as a less toxic substitute for cyclosporin against organ transplant rejection.
CONCLUSIONS
Although largely distinct and seemingly unrelated diseases, asthma and PAH have important pathological features in common, including inflammation, smooth muscle contraction and remodelling. These common features, although often unequally manifested in the two disorders, can be explained by one shared pathogenetic mechanism: activation of transcription factor NFAT. If this concept is validated, it could lead to the introduction of novel, more effective therapeutic strategies against both lung disorders, and possibly others. Although particularly notable in “severe asthma”, varying degrees of airway remodelling occur in human asthmatics 2, 24, and in a variety of experimental models of the disease in rodents. In at least some of these models, remodelling of the airways is accompanied by remodelling of smaller pulmonary arteries, validating the hypothesis of their similar pathogenesis. VIP emerges as a probable physiological inhibitor of the calcineurin–NFAT pathway, which explains why deletion of the VIP gene results in expression of both the asthma and PAH phenotypes 33, 34. As a modulator of calcineurin–NFAT signalling, VIP would be expected to be beneficial in both asthma and PAH 50. The peptide has shown airway-relaxant, anti-inflammatory and anti-proliferative actions on human airway smooth muscle cells, but no major clinical trials of VIP or related peptides have been reported in asthmatics 51. Administered by inhalation, VIP treatment resulted in remarkable improvement in pulmonary haemodynamics and exercise tolerance in IPAH patients 52. Confirmation of the effectiveness of VIP in suppressing calcineurin–NFAT signalling would have additional favourable therapeutic implications, particularly as a safer alternative to cyclosporin A in preventing organ transplant rejection.
Support statement
S.I. Said and L. Gonzalez Bosc were supported by National Institutes of Health grants HL-70212 and HL-88151, respectively. The study was supported by the Dept of Veterans Affairs Medical Center (Northport, NY, USA).
Statement of interest
None declared.
- Received June 19, 2009.
- Accepted October 8, 2009.
- © ERS Journals Ltd