





Abstract
Increased numbers of macrophages and neutrophils in the lung is a key feature of chronic obstructive pulmonary disease (COPD). The major neutrophil chemotactic agent in the airways of COPD patients is leukotriene (LT)B4 and is released by macrophages. The present study examines the role and mechanism of Ca2+ in platelet-activating factor (PAF)-stimulated LTB4 release from human lung macrophages.
Macrophages were isolated from lung tissue of subjects undergoing lung resection surgery and monocyte-derived macrophages (MDM) were obtained from nonsmokers, smokers without obstruction and COPD patients. Cells were stimulated with PAF and LTB4 release and [Ca2+]i was measured.
Lung macrophages and MDM released LTB4 following stimulation with PAF (mean effective concentration: 0.08±0.06 μM (n = 5) versus 0.17±0.12 μM (n = 17), respectively). Compared with MDM, lung macrophages released approximately eight-fold more LTB4. Neither smoking nor COPD altered MDM responses. PAF-stimulated LTB4 release was abrogated by ethylene glycol tetraacetic acid suggesting a role for extracellular Ca2+. This was substantiated by using store-operated channel blockers econazole, SK&F96365 and Gd3+. However, econazole and SK&F96365 were more effective in MDM than lung macrophages. Neither LOE908 nor nifedipine could attenuate this response.
These data suggest that platelet-activating factor-stimulated leukotriene B4 release from human lung macrophages is mediated, in part, by Ca2+ influx through receptor- but not voltage-operated Ca2+ channels.
Neutrophils and macrophages are key components of the innate immune system. These phagocytic cells recognise and remove pathogens and, thus, are the first defence mechanism against infection. However, these cells also release inflammatory mediators including cytokines and chemokines that, when dysregulated, may drive chronic inflammation. In the lung, such an accumulation of macrophages and neutrophils is observed in chronic obstructive pulmonary disease (COPD) [1]. COPD is predicted to become the third most common cause of death in the world by 2020 [2]. This is thought to reflect the increasing global epidemic of cigarette smoking, the most common risk factor leading to the development of this debilitating disease [1].
Macrophages are the predominant cell type found in bronchoalveolar lavage (BAL) fluid from patients with COPD but have been also identified in increased numbers within the lung parenchyma, particularly at the sites of alveolar destruction [3]. These macrophages possess the capacity to mediate many of the pathophysiological features of COPD and are considered a target for novel anti-inflammatory therapies. The neutrophil may also contribute to the pathophysiology of COPD as these cells release elastolytic enzymes and cytokines that promote lung destruction and inflammation [4]. Neutrophils tend to accumulate in the larger airways of patients with COPD and, as such, are found in increased numbers in induced sputum [5]. The reason for this is unclear, although the neutrophil chemoattractant, leukotriene (LT)B4, has been implicated in COPD [6]. LTB4 levels are increased in sputum from COPD patients and contribute to ∼30% of the neutrophilic chemotactic activity of sputum [6, 7]. In addition, LTB4 levels are increased in BAL fluid of ex-smokers with emphysema compared with both former and current smokers without emphysema [8]. The precise source of LTB4 in the airways is unknown; however, it is considered the predominant neutrophil chemoattractant released by human alveolar macrophages [9] and can be released following stimulation with ionophore and arachidonic acid [10]. Human alveolar macrophages respond to platelet-activating factor (PAF) by releasing superoxide anions and LTB4 [11, 12]. Notably, alveolar macrophages from smokers are more sensitive to PAF-stimulation with respect to superoxide production compared with cells from nonsmokers [11]. Since cigarette smoking is the major risk factor for the development of COPD, this suggests that smoking may sensitise macrophages to PAF-stimulation and heighten release of inflammatory mediators including LTB4. In addition, LTB4 receptors are up-regulated in COPD [13] supporting the hypothesis that modulation of the leukotriene pathways may be an effective therapeutic anti-inflammatory approach for this disease [14].
There is little information regarding the regulation of LTB4 production by macrophages in COPD; however, macrophages from asthmatic subjects release increased levels of LTB4 following stimulation with calcium ionophore compared with cells from nonasthmatic subjects [12]. PAF also stimulates [Ca2+]i in human alveolar macrophages [15] suggesting that control of Ca2+ influx in these cells could regulate LTB4 release and, thus, modulate neutrophilic inflammation. Regulation of [Ca2+]i is an important mechanism controlling migration and cellular activation in many cell types, although there is scant information regarding regulation of [Ca2+]i in human macrophages. LTB4 release from rat peritoneal macrophages is attenuated in the absence of extracellular Ca2+ further supporting the role of Ca2+ in modulating macrophage functions [16]. Identification of underlying mechanisms responsible for PAF-stimulated LTB4 release from human lung macrophages could facilitate the development of novel therapeutic agents capable of inhibiting macrophage activation and potentially attenuating neutrophillic inflammation in COPD. Therefore, the present study sought to investigate the role of Ca2+ in PAF-stimulated LTB4 release from human lung macrophages and monocyte-derived macrophages (MDM), and the contribution of receptor- versus voltage-operated Ca2+ channels in mediating this response.
METHODS
Subject selection
Subjects with stable COPD that fulfilled American Thoracic Society criteria [17] were recruited from the Royal Brompton Hospital (London, UK) and King Edward VII Hospital (Windsor, UK) outpatient departments, and a general practice database. Nonsmokers and smokers were recruited from patients who had been investigated previously for other reasons as well as volunteers. Demographic data for blood donors is presented in table 1⇓. There were no significant difference in the smoking history between smokers and COPD patients (table 1⇓). Lung tissue was obtained from patients undergoing lung resection surgery. All subjects gave written informed consent, and the study was approved by the Brompton, Harefield and National Heart and Lung Institute and East Berkshire Research Ethics Committee.
Demographic data of blood donors
Isolation of monocytes and MDM
Peripheral blood mononuclear cells (PBMC) were isolated from whole blood as previously described [18]. Monocytes were isolated from the PBMC fraction using the Monocyte Isolation kit II (Miltenyi Biotec, Woking, UK), and resuspended in MDM media (RPMI-1640 medium supplemented with 2 mM l-glutamine, 10,000 units·mL−1 penicillin, 10 mg·mL−1 streptomycin and 10% (volume/volume) foetal bovine serum (Invitrogen Ltd, Paisley, UK)) and seeded at 1×106 cells·mL−1. Cells were cultured in the presence of 2 ng·mL−1 granulocyte-macrophage colony-stimulating factor for 12 days.
Isolation and culture of human lung macrophages
Lung macrophages were isolated from resected lung tissue and adhered to tissue culture plates as described previously [19]. Cells were confirmed as >95% pure macrophages using CD68 immunocytochemistry as described previously [20].
Measurement of LTB4 production
MDM and lung tissue macrophages were pre-incubated with or without reference compounds SK&F96365, econazole, 2-aminoethocydiphenyl borate (2-APB) and gadolinium (Gd3+) in HBSS (or phosphate-free buffer for Gd3+) at 37°C for 3 min. The cells were subsequently incubated in the presence of PAF (3 μM) for 30 min. The calcium ionophore, ionomycin (1 μM), was used as a positive control and 2 mM ethylene glycol tetraacetic acid (EGTA) was used as the negative control. The supernatants were immediately harvested and stored at -80°C until analysed for LTB4 release using a commercially available enzyme immunoassay kit according to the manufacturer’s instructions (GE Healthcare, Amersham, UK). Each data point was measured in triplicate for the number of individual donors.
Measurement of [Ca2+]i using fluorescence
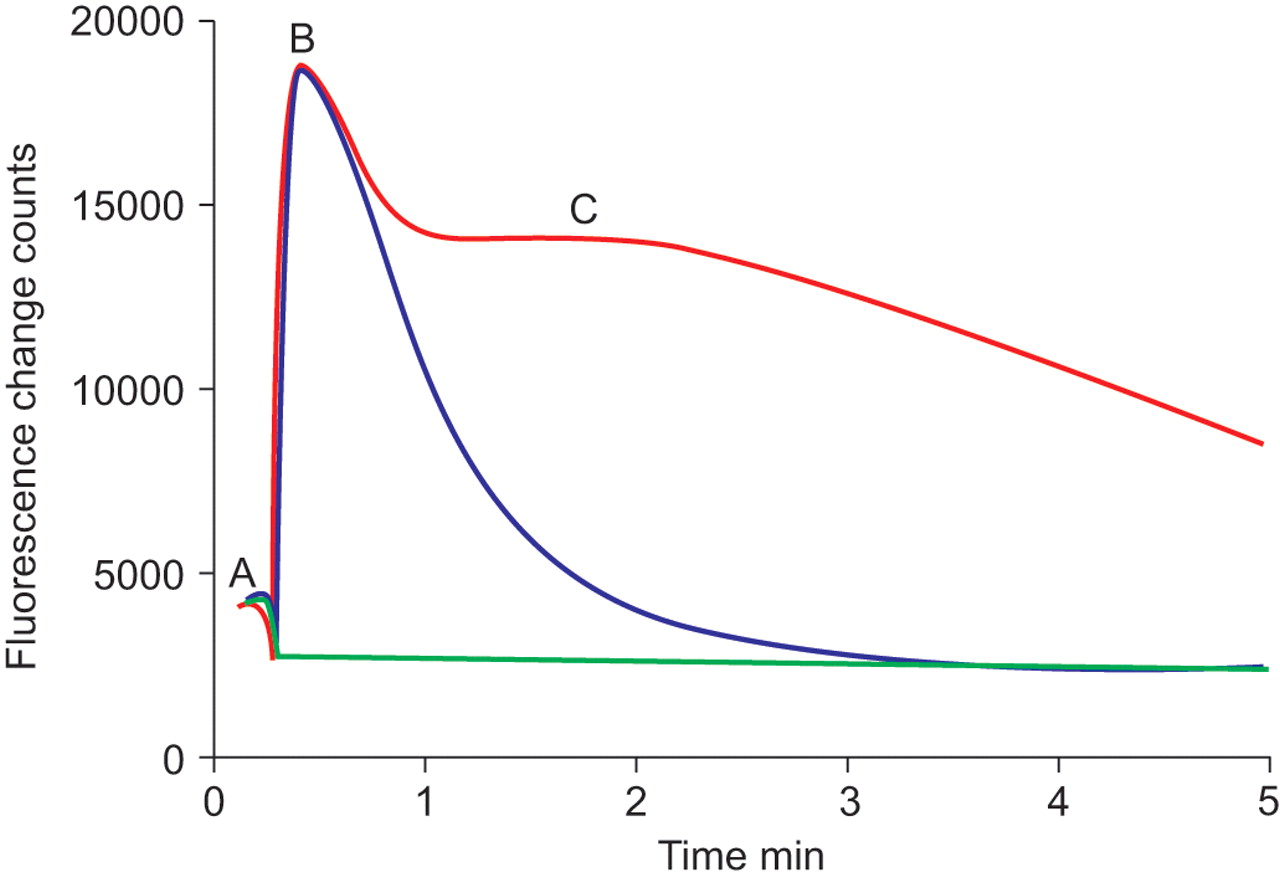
[Ca2+]i was measured using a fluorescence imaging plate reader (FLIPR; Molecular Devices Corporation, Sunnyclale, CA, USA). Culture medium was replaced with 100 μL of Fluo-4 (Invitrogen Ltd) loading medium containing 1 μM Fluo-4 acetoxymethyl ester, 205 mM probenecid, 20 mM hydroxyethyl piperazine ethane sulphonic acid (HEPES) and 100 μM brilliant black (ICN Biochemicals, Cambridge, UK). The plates were incubated at 37oC, 5% CO2 for 30 min prior to each experiment. Experiments were performed in HBSS assay buffer (HBSS containing 2.5 mM probenecid and 20 mM HEPES, containing 1 mM CaCl2 except for the studies using Gd3+, where phosphate-free buffer was used (140 mM NaCl, 5.4 mM KCl, 1.0 mM MgCl2, 1.2 mM CaCl2, 15 mM HEPES and 2.5 mM probenecid, pH 7.4)). The PAF receptor antagonist, PCA4248 [21] (Biomol, Exeter, UK) was added offline to the cells 30 min prior to the experiment. Compound solutions (SK&F96365, econazole, LOE908 (QuChem, Belfast, UK) [22], nifedipine and Gd3+) were added by injection and incubated with the cells for ∼2 min prior to the addition of PAF (1 μM). A representative FLIPR trace indicating how [Ca2+] was determined is presented in figure 1⇓. PAF stimulated [Ca2+]i release was calculated as:

Representative fluorescence imaging plate reader (FLIPR) traces of [Ca2+]i levels in lung macrophages from a single donor. Levels of [Ca2+]i were measured in lung macrophages following no stimulation (green line), 1 μM platelet-activating factor (PAF; red line) or PAF in the presence of 2 mM ethylene glycol tetraacetic acid (blue line). A: basal [Ca2+]i level in the lung macrophages; B: the peak of PAF-stimulated [Ca2+]i release; C: the Ca2+ influx response. FLIPR is manufactured by Molecular Devices Corporation (Sunnyclale, CA, USA).

To distinguish Ca2+ release from intracellular stores from Ca2+ influx, the effect of 2 mM EGTA was determined.
The percentage of the total PAF-induced Ca2+ response that contributed to Ca2+ influx was calculated as:

where AUC indicates area under the curve. The Ca2+ influx response inhibited by the ion channel blockers is expressed as the percentage of the total PAF-induced Ca2+ influx response, i.e.

Cell viability
Cell viability was determined by measuring reduction of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, to formazan by mitochrondial dehydrogenases, as described previously [23]. None of the treatments used in this study altered cell viability.
Statistical analysis
Data are presented as mean±sem. Comparisons between subject groups were performed using Kruskall–Wallis analysis. Differences were considered significant where p<0.05.
RESULTS
PAF-mediated LTB4 release by macrophages
Initial experiments demonstrated that PAF stimulation of macrophages had little effect on the release of CXCL8, interleukin-6 or matrix metalloproteinase-9 from these cells (data not shown), but stimulated the release of LTB4 from both lung macrophages and MDM significantly above baseline (p<0.05; fig. 2a⇓ and b). PAF was significantly more efficacious at stimulating LTB4 release from lung macrophages (p<0.01) compared with MDM, although there was no significant difference in the median effective concentration (EC50) values between lung macrophages and MDM (0.08±0.06 μM (n = 5) versus 0.17±0.12 μM (n = 17), respectively). Despite differences in the demographics of the blood donors (table 1⇑), when the data from MDM were separated into nonsmokers, smokers and COPD patients, there were no differences in PAF-stimulated LTB4 release (EC50 0.40±0.37 μM, 0.11±0.08 μM and 0.30±0.01 μM for five nonsmokers, six smokers and six COPD patients, respectively). However, there were no significant differences in the levels of LTB4 released with 0.1–10 μM PAF due to the variation in responses from each of the individual human donors.

Effect of platelet-activating factor (PAF) and ionomycin on release of leukotriene (LT)B4 by lung macrophages and monocyte-derived macrophages (MDM). a) Lung macrophages (n = 5 donors) and b) MDM (n = 17 donors) were stimulated with increasing concentrations of PAF in the absence (▪) or presence of 2 mM ethylene glycol tetraacetic acid (▴) and the levels of released LTB4 measured by enzyme immunoassay. c) Lung macrophages (•) and MDM (▪) were stimulated with increasing concentrations of ionomycin. The media was harvested and LTB4 release was measured. Data are presented as mean±sem.
PAF-stimulated LTB4 release from both lung macrophages and MDM was reduced in the presence of 2 mM EGTA, indicating a dependency on Ca2+ influx (fig. 2a⇑ and b). The nature of this response was examined using the PAF receptor antagonist PCA4248 (10 μM), which inhibited the release of LTB4 induced by 3 μM PAF by >95% (data not shown). The enhanced release of LTB4 by lung macrophages compared with MDM was confirmed using calcium ionophore (fig. 2c⇑).
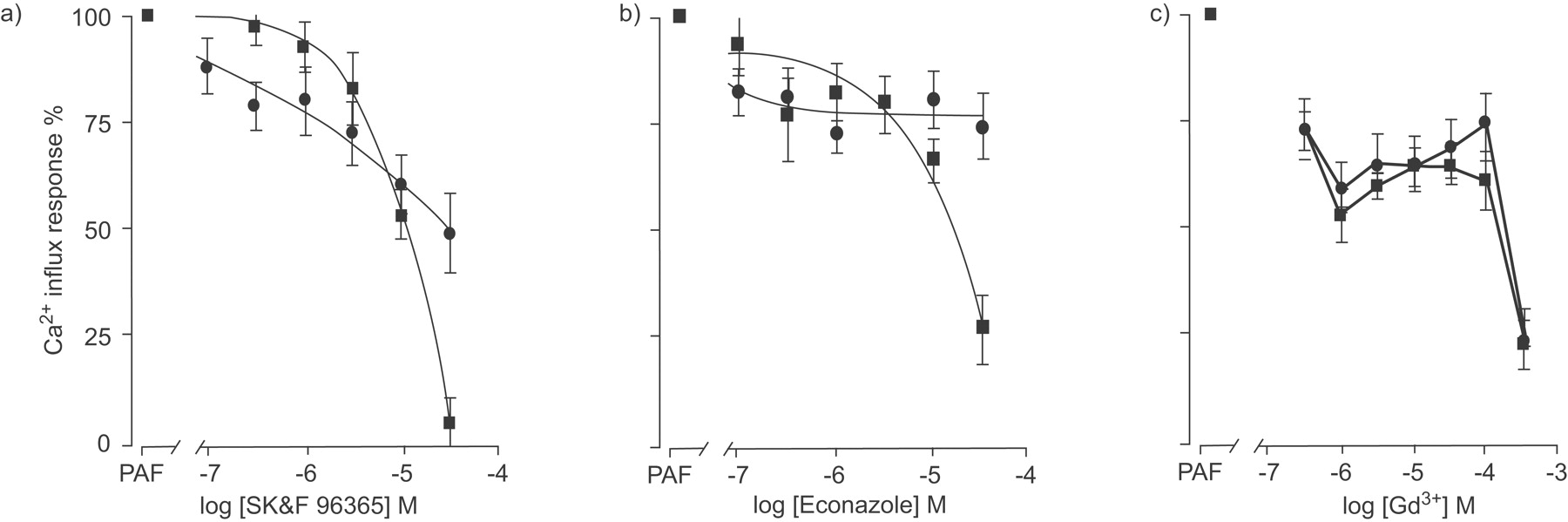
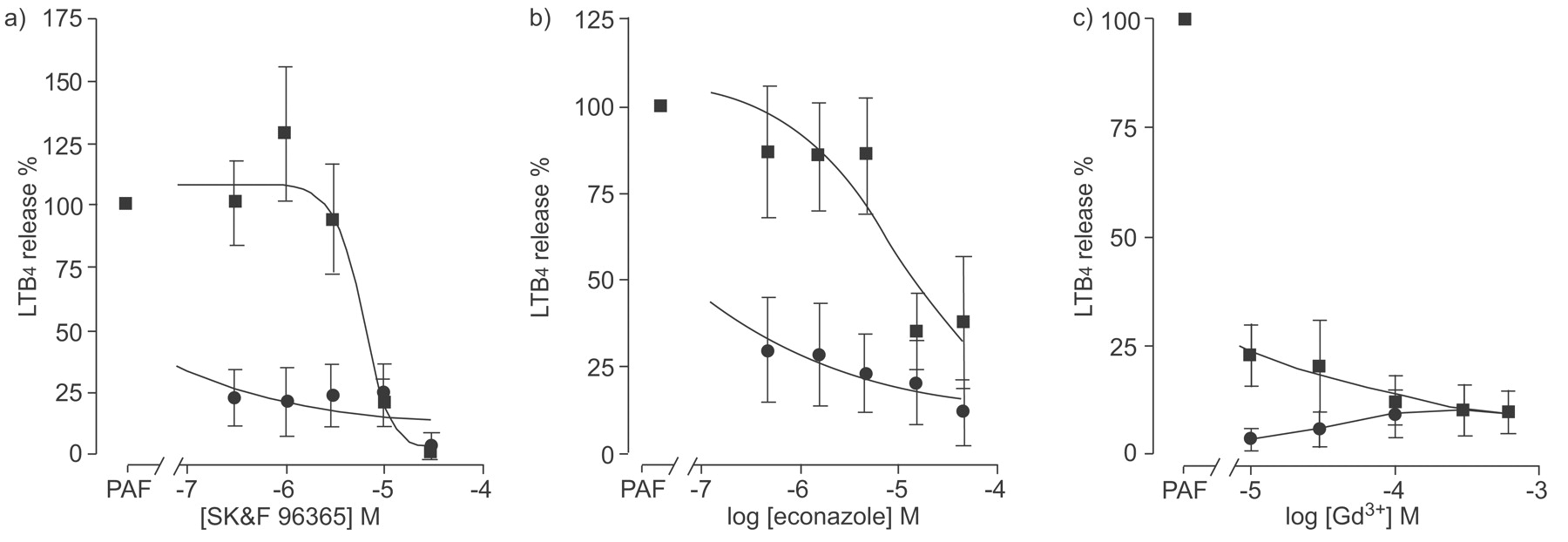
To investigate these differences further, the effects of various pharmacological ion channel blockers on 3 μM PAF-induced LTB4 release from both lung macrophages and MDM were also determined. PAF-induced release of LTB4 from lung macrophages was inhibited by SK&F96365, econazole and Gd3+ (fig. 3⇓). These responses contrasted with those seen when the same channel blockers were used to examine PAF-induced LTB4 release by MDM (fig. 3⇓). SK&F96365 and econazole concentration-dependently inhibited PAF-stimulated LTB4 release from MDM (EC50 5.4±1.1 μM (n = 19) and 2.2±1.0 μM (n = 17) for SK&F96365 and econazole, respectively; fig. 3a⇓ and b), which differed from the responses seen in lung macrophages where it was not possible to calculate EC50 values. In contrast, the responses of both MDM and lung macrophages to the addition of Gd3+ prior to stimulation with PAF was similar for both cell types (EC50 <10 μM (n = 12) and 137±122 μM (n = 15) for lung macrophages and MDM, respectively; fig. 3c⇓).

Effect of channel blockers, SK&F96365, econazole, and gadolinium (Gd3+) on platelet-activating factor (PAF)-induced leukotriene (LT)B4 release by lung macrophages and monocyte-derived macrophages (MDM). Lung macrophages (•; n = 5 donors) or MDM (▪; n = 16 donors) were stimulated with 1 μM PAF in the presence of a) SK&F96365, b) econazole or c) Gd3+. Media was harvested and LTB4 release was measured. Data are presented as mean±sem.
Ca2+ influx into macrophages
PAF demonstrated the largest sustained Ca2+ response in both lung macrophages (fig. 4a⇓) and MDM (fig. 4b) when compared with other chemotactic factors including f-Met-Leu-Phe and C5a. The inhibitor of sarco-endoplasmic reticulum Ca2+-adenosine triphosphatases, thapsigargin, stimulated [Ca2+]i in lung tissue macrophages (fig. 4a⇓) and MDM (fig. 4b⇓). The sustained Ca2+ response observed following stimulation of macrophages with PAF was not due to a feedback mechanism via the action of LTB4, since stimulation of the cells with LTB4 did not directly support a sustained Ca2+ response (fig. 4c⇓). Sustained increases in [Ca2+]i observed following stimulation of both macrophages and MDM with PAF were not apparent when the cells were stimulated with chemokines (fig. 5⇓). In both MDM and lung macrophages, increases in [Ca2+]i were observed following stimulation with CXCL12, CCL5 and CCL3 (fig. 5a⇓ and b) but was much reduced compared with the response of these cells to PAF (fig. 5c⇓). In contrast, CXCL1, CCL2, CXCL9, CXCL10 or CXCL11 failed to increase [Ca2+]i in both lung macrophages and MDM (data not shown). PAF increased [Ca2+]i in lung macrophages in a concentration-dependent manner with an EC50 value of 37.4±5.8 nM (n = 3) and was confirmed as a PAF receptor-mediated effect as the PAF receptor antagonist, PCA4248, concentration-dependently inhibited this response (inhibitor concentration of 50% (IC50) value of 8.8±1.9 μM (n = 3)). Similar responses were seen with MDM following PAF stimulation generating a concentration-dependent increase in [Ca2+]i with an EC50 value of 13.6±5.4 nM (n = 3) and PCA4248 inhibiting this effect with an IC50 value of 14.2±2.6 μM (n = 3).

Representative fluorescence imaging plate reader (FLIPR) traces of [Ca2+]i levels in a) lung macrophages and b) monocyte-derived macrophages (MDM) following stimulation with buffer, platelet-activating factor (PAF; 1 μM), C5a (100 nM), f-Met-Leu-Phe (fMLP; 1 μM) and thapsigargin (1 μM). c) The levels of [Ca2+]i were measured in lung macrophages following stimulation with leukotriene (LT)B4. Traces are representative of four to 12 individual experiments. FLIPR is manufactured by Molecular Devices Corporation (Sunnydale, CA, USA).

Representative fluorescence imaging plate reader (FLIPR) traces of [Ca2+]i levels in lung macrophages and monocyte-derived macrophages (MDM) following stimulation with chemokines. Levels of [Ca2+]i were measured in a) MDM and b) lung macrophages following stimulation with CCL3 (100 nM), CCL5 (200 ng·mL−1) and CXCL12 (100 nM). c) Comparison of platelet-activating factor (PAF)-induced [Ca2+]i with that from CXCL12, CCL3 and CCL5 in lung macrophages. Traces are representative of two to 12 individual experiments. FLIPR is manufactured by Molecular Devices Corporation (Sunnydale, CA, USA).
To delineate Ca2+ release from intracellular stores from Ca2+ influx, the effect of EGTA on PAF-dependent [Ca2+]i was determined and showed that much of the PAF-mediated effect on [Ca2+]i was Ca2+ influx dependent in both lung macrophages and MDM (63.9±2.9% (n = 13) and 73.5±4.9% (n = 4) inhibition, respectively). To examine this response further, the effect of various Ca2+ influx blockers on the PAF-induced Ca2+ influx were determined in both lung macrophages and MDM (fig. 6⇓). SK&F96365 (fig. 7a⇓) and econazole (fig. 7b⇓) only partially inhibited the PAF-stimulated Ca2+ influx response with maximum inhibition of approximately 50% and 25%, respectively, in lung macrophages with an EC50 value of 1.7±0.8 μM (n = 12) for SK&F96365. In contrast to the effect of SK&F96365 and econazole on lung macrophages, PAF-induced Ca2+ influx was maximally inhibited by approximately 90% and 70% in MDM (fig. 7a⇓ and b) with EC50 values of 11.1±1.8 μM (n = 10) and 7.0±2.8 μM (n = 10), respectively. The responses of both lung macrophages and MDM to the trivalent cation channel blocker, Gd3+, were similar with significant inhibition of Ca2+ influx (EC50: 32.2±15.4 μM (n = 12) and 12.1±7.3 μM (n = 10) for lung macrophages and MDM, respectively; fig. 7c⇓).

Representative fluorescence imaging plate reader (FLIPR) traces of [Ca2+]i levels examining the effect of channel blockers on platelet-activating factor (PAF)-induced Ca2+ influx by lung macrophages. Levels of [Ca2+]i were measured in lung macrophages following a) pre-incubation with the channel blockers SK&F96365 (10 μM), econazole (30 μM) and gadolinium (Gd3+; 300 μM) and stimulation with PAF (3 μM), b) pre-incubation with nifedipine (10 μM) or c) pre-incubation with 2-aminoethocydiphenyl borate (2-APB) prior to stimulation with PAF. Traces are representative of three to 11 individual experiments. EGTA: ethylene glycol tetraacetic acid. FLIPR is manufactured by Molecular Devices Corporation (Sunnydale, CA, USA).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of channel blockers, SK&F96365, econazole and Gd3+ on platelet-activating factor (PAF)-induced Ca2+ release. Lung macrophages (•; n = 11 donors) and monocyte-derived macrophages (▪; n = 10 donors) were stimulated with 1 μM PAF in the presence of a) SK&F96365, b) econazole or c) gadolinium (Gd3+). [Ca2+]i was measured using fluorescence imaging plate reader (FLIPR). Data are presented as mean±sem. FLIPR is manufactured by Molecular Devices Corporation (Sunnydale, CA, USA).
The responses of lung macrophages and MDM to all three channel blockers indicated that PAF-induced LTB4 release by lung macrophages was more sensitive to inhibition when compared with the effect of these compounds on Ca2+ influx. However, the inhibitory effects of the channel blockers econazole and SK&F96365 for both [Ca2+]i and LTB4 release were similar in MDM. Although, in these cells, the inhibitory effect of Gd3+ was ∼30-fold greater for LTB4 release when compared with Ca2+ influx. These data prompted further analysis of the PAF-induced Ca2+ response in lung macrophages where the selective l-type voltage-operated Ca2+ channel blocker, nifedipine (fig. 6b⇑), and the nonselective cation channel blocker, LOE908, were ineffective (data not shown). In addition, 2-APB, an inhibitor of store-operated channels [24], also blocked PAF-induced Ca2+ influx (EC50 5.8±0.3 μM (n = 5); fig. 6c⇑) and release of PAF-stimulated LTB4 from these cells (PAF 1.3±0.3 ng·mL−1 versus 50 μM 2-APB 0.2±0.06 ng·mL−1, n = 4, p<0.05). Similarly, Ni2+ also abrogated Ca2+ influx in lung macrophages with an EC50 value of 502.3±141 μM (n = 5; data not shown). Taken together, these data suggest that PAF-stimulated Ca2+ influx is mediated via receptor-operated and not l-type voltage-operated calcium channels in macrophages.
DISCUSSION
PAF is produced in the airways during inflammation and may mediate macrophage activation, including production of LTB4, a potent neutrophil chemotactic agent [12]. PAF exposure of both lung macrophages and MDM stimulated the largest response for both LTB4 release and Ca2+ influx. Furthermore, the present study confirmed that release of LTB4 from both lung macrophages and MDM is dependent upon Ca2+ influx similar to that reported for human neutrophils [25]. By contrast, lung macrophages and MDM did not respond to a number of chemokines including CXCL9, CXCL10 and CXCL11. Similarly, stimulation of CXCR2 and CCR2 also failed to elicit Ca2+ signals in these cells. However, both lung macrophages and MDM responded to CXCL12, CCL3 and CCL5. However, these responses suggest that receptor-operated channels in the macrophage membrane are agonist selective as the effects of chemokines are distinct from those observed when macrophages were stimulated with agonists, such as PAF and C5a.
The concentrations of PAF that stimulated release of LTB4 from both lung macrophages and MDM was similar to that reported to elicit superoxide production by alveolar macrophages (EC50 ∼0.1 μM) [11]. In contrast to superoxide production from alveolar macrophages [11], PAF-stimulated release of LTB4 from MDM was not altered by smoking or disease status, but does support data obtained from alveolar macrophages from smokers and COPD patients whereby these cells release similar amounts of LTB4 under resting conditions (smokers: 244±107 pg·mL−1; COPD: 137±61 pg·mL−1) and following stimulation with lipopolysaccharide (LPS; smokers: 342±99 pg·mL−1; COPD: 226±94 pg·mL−1) [26].
Although both lung macrophages and MDM displayed similar concentration-dependent responses to PAF stimulation, the magnitude of the LTB4 response was greater in lung macrophages compared with MDMs. This may reflect differential expression of PAF-acetyl hydrolase (PAF-AH) by these cells since PAF-AH catabolises PAF. However, as lung macrophages stimulated with ionomycin also release increased levels of LTB4 compared with MDM, differences in the regulation of the arachidonic acid metabolic pathways could also account for these results. Alternatively, these data suggest differential regulation of store-operated calcium channels since the effects of receptor-operated channel blockers SK&F96365 and econazole differed between lung macrophages and MDM.
The relationship between changes in [Ca2+]i and release of LTB4 in both lung macrophages and MDM is complex and may represent differential coupling of the PAF response. PAF stimulates Ca2+ influx in macrophages [27]; however, the concentration of PAF required to increase [Ca2+]i in the present study is ∼10–100-fold greater than that reported for hamster and guinea pig macrophages [28], but is similar to that reported for human MDM (EC50 ∼10nM) [29]. Ca2+ influxes observed in lung macrophages following PAF stimulation were relatively resistant to econazole but were inhibited by ∼50% with SK&F96365. Both econazole and SK&F96365 attenuate receptor mediated calcium entry into cells with reported IC50 values of ∼3 μM and 10 μM, respectively [30, 31], and were more effective at blocking Ca2+ influx in MDM when compared with lung macrophages, although these channel blockers are not selective [32]. These data also strongly suggest that PAF-stimulated LTB4 release from lung macrophages is not solely due to Ca2+ influx but may be mediated by other channels since release of LTB4 by lung macrophages was more sensitive to inhibition by both econazole and SK&F96365 when compared with Ca2+ influx. However, the inhibitory effect of Gd3+ in both MDM and lung macrophages was ∼30-fold greater for LTB4 release when compared to the effect on Ca2+ influx. Gd3+ has selectivity for transient receptor potential (TRP) channels in the concentration range 100–300 μM, where this compound showed efficacy for PAF mediated Ca2+ influx. Further examination of PAF-induced Ca2+ influx in lung macrophages demonstrated that the nonselective cation channel blocker, LOE908, and the selective l-type voltage-operated Ca2+ channel blocker, nifedipine, were ineffective, suggesting that this response is mediated via a receptor-operated channel and is consistent with previous observations whereby no voltage-dependent Ca2+ channels have been detected electrophysiologically in macrophages [33]. Therefore, it is possible that a TRP-type channel may be important in regulating Ca2+ in these cells.
The differential sensitivity of lung macrophages to channel blockers and the increased levels of LTB4 release from these cells compared with MDM suggests that differentiation or exposure of macrophages in a lung environment alters the expression of ion channels or “primes” these cells to produce more LTB4. Exposure of alveolar macrophages from smokers to LPS potentiates release of LTB4 when compared with cells from ex-smokers [34]; therefore, priming agents in the lung may potentiate release of LTB4 from lung macrophages. The precise nature of these agents is not known; however, lung macrophages were obtained from patients undergoing surgery and included smokers and ex-smokers.
These data suggest that platelet-activating factor stimulation of human macrophages leads to leukotriene B4 release by a mechanism which is dependent, in part, on extracellular Ca2+ influx, with receptor-, but not voltage-operated calcium channels responsible for mediating this effect. The differences in the pharmacological profiles of monocyte-derived macrophages and lung macrophages suggest that monocyte-derived macrophages are not good surrogates for lung macrophages with respect to Ca2+ homeostasis and may reflect an alternative differentiation pathway for macrophages in the lung, or represent a primed population of macrophages. However, understanding of the regulation of macrophage functions and the role of ion channels in these responses may provide novel therapeutic opportunities for the treatment of inflammatory lung diseases, such as chronic obstructive pulmonary disease.
Statement of interest
Statements of interest for P.J. Barnes and L.E. Donnelly can be found at www.erj.ersjournals.com/misc/statements.dtl
- Received August 22, 2008.
- Accepted December 5, 2008.
- © ERS Journals Ltd
References










