





Abstract
The cholinergic nervous system can inhibit the systemic inflammation accompanying sepsis by virtue of a specific action of acetylcholine on α7 cholinergic receptors. The current authors sought to determine the effect of nicotine, an α7 cholinergic receptor agonist, on the host response to pneumonia caused by Streptococcus pneumoniae.
Mice were intranasally infected with S. pneumoniae and treated with nicotine or saline intraperitoneally using a treatment schedule shown to improve host defence against abdominal sepsis.
Nicotine treatment was associated with a transiently enhanced growth of S. pneumoniae, as indicated by higher bacterial loads in both lungs and blood at 24 h after infection. At 48 h after infection, bacterial burdens had increased in both treatment groups and differences were no longer present. Remarkably, mice treated with nicotine showed enhanced lung inflammation at 24 h after infection. Moreover, both lung and plasma concentrations of the pro-inflammatory cytokines tumour necrosis factor-α and interferon-γ were higher in nicotine-treated animals at this time-point. Additional studies examining the effect of nicotine on the immediate (4-h) inflammatory response to S. pneumoniae did not reveal an anti-inflammatory effect of nicotine either.
The present data suggest that nicotine transiently impairs host defence in pneumococcal pneumonia.
The cholinergic nervous system can regulate inflammation via its principal neurotransmitter, acetylcholine 1. In the so-called cholinergic anti-inflammatory pathway, enhanced efferent activity of parasympathetic nerve endings results in the release of acetylcholine, which, by a specific action on α7 nicotinic cholinergic receptors on macrophages, inhibits pro-inflammatory cytokine production 2. Disruption of this pathway by surgical division of the vagus nerve led to enhanced release of tumour necrosis factor (TNF)-α and accelerated the development of hypotensive shock after i.v. injection of lipopolysaccharide (LPS) into rats 3. Conversely, electrical stimulation of the efferent vagus nerve prevented the development of shock and attenuated the release of TNF-α in endotoxemic rats 3, 4. Moreover, stimulation of α7 cholinergic receptors by specific agonists, such as nicotine or 3-(2,4-dimethoxybenzylidene) anabaseine (GTS-21), attenuated TNF-α release and improved survival in mice challenged with LPS 5–7 and in mice with abdominal sepsis induced by cecal ligation and puncture (CLP) 6, 7.
Recent evidence suggests that stimulation of α7 cholinergic receptors in the lung inhibits local inflammation. Alveolar macrophages and respiratory epithelial cells were reported to express α7 cholinergic receptors in the normal lung, whereas, in lungs with acid aspiration-induced injury, expression of α7 cholinergic receptors was also detected on infiltrating neutrophils 8. In addition, systemic administration of nicotine, choline or the specific α7 agonist PNU-282987 attenuated acid aspiration-induced lung injury and inflammation, as reflected by a reduction in lung vascular permeability, neutrophil influx into bronchoalveolar lavage fluid (BALF) and local TNF-α concentrations 8. Moreover, it was shown that intrapulmonary delivery of the α7 agonist GTS-21 attenuated TNF-α release into BALF of mice exposed to LPS via the airways 9. To date, knowledge of the effect of cholinergic stimulation during lung infection is limited to one study, in which nicotine administration to mice was associated with enhanced viral loads during experimental influenza infection 10. The current authors sought to determine the effect of nicotine on host defence against bacterial pneumonia. Such knowledge is important, considering that pneumonia has a considerable impact on healthcare and is the leading cause of sepsis 11, 12.
MATERIALS AND METHODS
Mice
Pathogen-free 9-week-old female C57BL/6 mice were purchased from Harlan (Horst, the Netherlands). The Institutional Animal Care and Use Committee of the Academic Medical Center (Amsterdam, the Netherlands) approved all experiments. At the start of the experiments, mice were 10 weeks old.
Induction of pneumonia and design
Pneumonia was induced as previously described 13–15. Briefly, Streptococcus pneumoniae serotype 3 (ATCC 6303; American Type Culture Collection, Rockville, MD, USA) were grown for 6 h to mid-logarithmic phase at 37°C using Todd–Hewitt broth (Difco, Detroit, MI, USA), harvested by centrifugation at 1500×g for 10 min and washed twice in sterile isotonic saline. Bacteria were resuspended in sterile isotonic saline at a concentration of 1×106 colony-forming units (CFU)·mL−1, as determined by plating serial 10-fold dilutions on blood agar plates. After preparation of the inocula, mice were lightly anaesthetised by inhalation of 2% isoflurane (Abbott Laboratories Ltd, Queenborough, UK) in 2 L of O2, and 50 μL of the bacterial suspension (containing 5×104 CFU S. pneumoniae) was inoculated intranasally. Mice received an i.p. injection (total volume 200 μL) with either vehicle (sterile normal saline) or nicotine (Sigma-Aldrich Co., St Louis, MO, USA) at a dose of 400 µg·kg−1 starting directly after infection and with 8-h intervals thereafter until the end of each experiment; this treatment schedule was previously shown to protect mice from death induced by either LPS or CLP-induced sepsis 7.
Sample harvesting and preparation
Mice were anaesthetised 4, 24 or 48 h after induction of pneumonia with Hypnorm® (Janssen Pharmaceutica, Beerse, Belgium; active ingredients fentanyl citrate and fluanisone) and midazolam (Roche, Meidrecht, the Netherlands) and sacrificed by bleeding out the vena cava inferior. Blood was collected in EDTA-containing tubes. Whole lungs were harvested and homogenised in four volumes of sterile saline using a tissue homogeniser (Biospec Products, Bartlesville, OK, USA). CFU numbers were determined in lungs and blood from serial dilutions plated on blood agar plates and incubated at 37°C for 16 h before colonies were counted. For cytokine measurements, lung homogenates were diluted with an equal volume of lysis buffer (pH 7.4) containing 300 mM NaCl, 30 mM Tris, 2 mM MgCl2, 2 mM CaCl2, 1% Triton X-100, 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride, EDTA, pepstatin A and leupeptin (all from MP Biomedicals, Solon, OH, USA; concentrations in accordance with the manufacturer’s recommendations) and incubated for 30 min. Homogenates were centrifuged at 1500×g at 4°C for 15 min, and supernatants were stored at -20°C until assays were performed.
Histology
Lungs for histology were prepared and analysed by a pathologist (S. Florquin) who was blinded with respect to treatment groups, as previously described 13–15. The parameters bronchitis, oedema, interstitial inflammation, intra-alveolar inflammation, pleuritis and endothelialitis were graded on a scale of 0 to 4, with 0 as “absent” and 4 as “severe”. The total “lung inflammation score” was expressed as the sum of the scores for each parameter, the maximum being 24. Additionally, the percentage of inflammation was determined. Granulocyte staining was performed using fluorescein isothiocyanate-labelled rat anti-mouse Ly-6G monoclonal antibody (mAb; Pharmingen, San Diego, CA, USA) exactly as previously described 14.
Assays
TNF-α, interleukin (IL)-6, interferon (IFN)-γ, IL-10 and monocyte chemoattractant protein (MCP)-1 levels were determined using a commercially available cytometric beads array multiplex assay (BD Biosciences, San Jose, CA, USA) in accordance with the manufacturer’s recommendations. Macrophage inflammatory protein (MIP)-2 and keratinocyte chemoattractant (KC) were measured by ELISA (R&D Systems, Abingdon, UK).
Statistical analysis
All values are presented as mean±sem. Differences between groups were analysed by Mann–Whitney U-tests. Correlations between bacterial loads and lung cytokine concentrations were calculated using the Spearman rho test. Values of p<0.05 were considered to be statistically significant.
RESULTS
Nicotine transiently impairs antibacterial defence
To obtain a first insight into the impact of nicotine on host defence during pneumonia, mice were infected with S. pneumoniae and concurrently treated with either nicotine (400 µg·kg−1) or normal saline every 8 h 7. In the first experiments, mice were killed 24 or 48 h after infection and bacterial loads were determined in whole lungs and blood (fig. 1⇓). Nicotine treatment was associated with significantly higher bacterial burdens in both lungs and blood at 24 h post-infection (both p<0.01 versus saline). Bacterial loads increased in both treatment groups thereafter and no differences existed at 48 h (fig. 1⇓).

Nicotine treatment transiently enhanced bacterial outgrowth in a) lung and b) blood. Mice were intranasally infected with Streptococcus pneumoniae and treated with either nicotine (400 µg·kg−1; □) or saline (▓) every 8 h until sacrifice at 24 or 48 h. Data represent mean±sem of eight mice per group at each time-point. **: p<0.01.
Impact of nicotine on lung inflammation
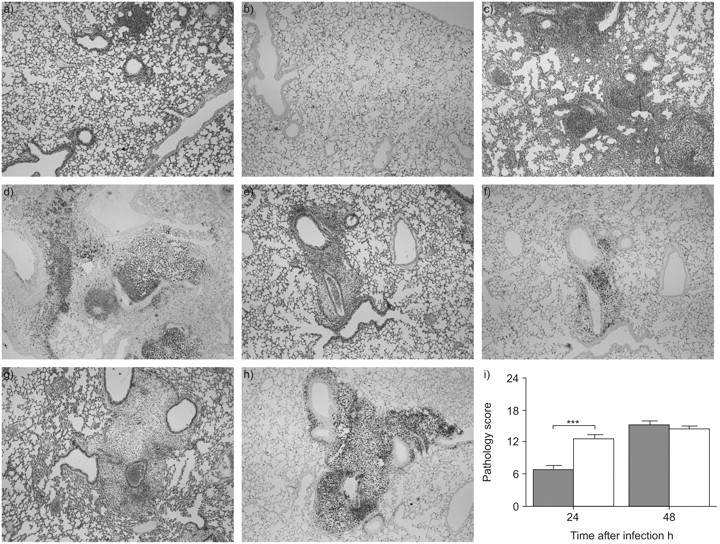
Nicotine and other α7 cholinergic agonists have been reported to exert anti-inflammatory effects in models of sterile lung injury 8, 9. In theory such anti-inflammatory effects could impair antibacterial defence, considering that a certain extent of inflammation is required for an adequate innate immune response to respiratory pathogens 16, 17. Therefore, the effect of nicotine on the pulmonary inflammation accompanying pneumonia was determined by semiquantitative analysis of lung histopathology (fig. 2⇓). Nicotine treatment was associated with increased lung inflammation at 24 h after induction of pneumonia, as reflected by an increased pathology score at this time-point (p<0.001 versus saline). The enhanced inflammatory response in nicotine-treated animals was further illustrated by a strongly increased neutrophil recruitment into lung tissue, as shown by Ly-6G staining for neutrophils (fig. 2b⇓ and d). At 48 h after infection, pathology scores no longer differed between groups. Since CXC chemokines have been implicated in the recruitment of neutrophils to sites of inflammation 18, the local concentrations of MIP-2 and KC were determined in whole lung homogenates. Nicotine did not influence pulmonary MIP-2 or KC levels at 24 h after infection. At 48 h, KC levels were lower in nicotine-treated mice (p<0.05 versus saline), whereas MIP-2 levels were similar in both groups (fig. 3⇓).

Nicotine transiently enhanced pneumonia-induced lung pathology. Mice were intranasally infected with Streptococcus pneumoniae and treated with either nicotine (400 µg·kg−1) or saline every 8 h until sacrifice at 24 or 48 h. Representative lung slides of saline-treated (a–d) and nicotine-treated (e–h) mice 24 h (a, b, e and f) and 48 h (c, d, g and h) after infection. Haematoxylin and eosin staining (a, c, e and g) and Ly-6G granulocyte staining (b, d, f and h) were performed. All pictures were taken at 4× magnification and are representative for eight mice per group at each time-point. i) Semiquantitative pathology scores (determined by a scoring system as described in the Methods section), 24 and 48 h after infection. Data represent mean±sem of eight mice per group at each time-point. ▓: saline-treated mice; □: nicotine-treated mice. ***: p<0.001.

Lung chemokine levels. Mice were intranasally infected with Streptococcus pneumoniae and treated with either nicotine (400 µg·kg−1; □) or saline (▓) every 8 h until sacrifice at 24 or 48 h. a) Macrophage inflammatory protein (MIP)-2 and b) keratinocyte chemoattractant (KC) levels were measured by ELISA. Data represent mean±sem of eight mice per group at each time-point. *: p<0.05.
Nicotine increases lung and plasma cytokine levels
To obtain a further insight into the effect of nicotine on the host inflammatory response during pneumococcal pneumonia, local and systemic cytokine concentrations were measured at 24 and 48 h after infection (fig. 4⇓). Nicotine treatment was associated with higher lung levels of TNF-α and IFN-γ, and higher plasma levels of TNF-α, IFN-γ and MCP-1 at 24 h after infection (all p<0.05 versus saline). At 48 h, neither lung nor plasma concentrations of these mediators differed between groups. The pulmonary and plasma levels of IL-6 and IL-10 were similar in both groups at both time-points. To evaluate whether lung cytokine levels were proportionate to the bacterial load, the correlations between pulmonary bacterial loads and cytokine levels 24 h after infection were calculated. Positive correlations were found between bacterial load and TNF-α (r = 0.71, p<0.005), IL-6 (r = 0.63, p<0.01) and IFN-γ (r = 0.74, p = 0.001); correlations between bacterial loads and MCP-1 or IL-10 levels were not significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lung (a–e) and plasma (f–j) levels of tumour necrosis factor (TNF)-α (a and f), interferon (IFN)-γ (b and g), interleukin (IL)-6 (c and h), IL-10 (d and i) and monocyte chemoattractant protein (MCP)-1 (e and j). Mice were intranasally infected with Streptococcus pneumoniae and treated with either nicotine (400 µg·kg−1; □) or saline (▓) every 8 h until sacrifice at 24 or 48 h. Data represent mean±sem of eight mice per group at each time-point. - - - -: detection limit. *: p<0.05; **: p<0.01.
Impact of nicotine on the early immune response
The enhanced cytokine response at 24 h post-infection in nicotine-treated mice could have been the result of the higher bacterial loads in these animals, providing a more potent pro-inflammatory stimulus. Therefore, the current authors wished to evaluate the influence of nicotine on the early inflammatory response to pneumonia. Mice were intranasally infected with S. pneumoniae, treated immediately thereafter with either nicotine (400 µg·kg−1) or saline, and lungs and blood were obtained 4 h later. At this early time-point, bacterial loads in lungs did not differ between groups (table 1⇓), whereas blood cultures remained sterile (data not shown). Lung concentrations of cytokines and chemokines did not differ between groups, with the exception of IL-6 levels, which were higher in nicotine-treated animals (p<0.05 versus saline; table 1⇓). Please note that overall cytokine levels were low in both groups. IFN-γ levels were below detection level. Plasma cytokine levels were either very low or undetectable in both nicotine- and saline-treated animals and not different between groups (data not shown).
Effect of nicotine on the very early(4-h) lung host response to pneumococcal pneumonia
DISCUSSION
The cholinergic anti-inflammatory pathway, mediated by the vagus nerve and α7 cholinergic receptors, has been implicated as a neuronal feedback system that serves to limit inflammatory responses 1. It has recently been demonstrated that chemical stimulation of α7 cholinergic receptors in the pulmonary compartment results in inhibition of sterile lung inflammation induced by local instillation of either LPS or acid 8, 9. The current authors sought to determine the effect of nicotine on the host response to respiratory tract infection caused by the most common causative pathogen in community-acquired pneumonia, S. pneumoniae. The main finding was that nicotine transiently impairs host defence during pneumococcal pneumonia as reflected by higher bacterial loads in lungs and blood in mice treated with nicotine, 24 h after induction of pneumonia. The enhanced growth of pneumococci was accompanied by increased inflammation in the lungs, as indicated by histopathology and cytokine levels.
Several lines of evidence indicate that stimulation of the vagus nerve and/or pharmacological α7 cholinergic receptor agonists may be a useful strategy in the treatment of the severe inflammation accompanying sepsis. First, electrical stimulation of the efferent vagus nerve prevented the development of shock and attenuated the release of TNF-α in endotoxemic rats 3, 4. Secondly, both electrical stimulation of the vagus nerve and stimulation of α7 cholinergic receptors by specific agonists diminished systemic inflammation and improved the outcome of mice with polymicrobial abdominal sepsis 6, 7, 19. Thus far, knowledge of the impact of stimulation of α7 cholinergic receptors in pneumonia has been highly limited. In light of the fact that pneumonia is the most common cause of sepsis 11, such knowledge is of great relevance for further exploration of manipulation of the cholinergic anti-inflammatory pathway as a potential novel therapeutic strategy in sepsis. In this respect, pneumonia caused by S. pneumoniae is especially important. S. pneumoniae is the most prevalent micro-organism in community-acquired pneumonia, responsible for more than half a million cases each year in the USA alone, bearing a fatality rate of 5–7% 20. Bacteraemia with S. pneumoniae originates, in almost 90% of cases, from the lungs. In addition, in recent sepsis trials, the pneumococcus was an important causative pathogen, especially in the context of pneumonia 21.
Recent studies have shown that α7 cholinergic receptors are expressed in the lung and that stimulation of these receptors by various treatments attenuates lung inflammation and injury 8, 9. In one of these investigations nicotine, given as a single dose of 3.5 mg·kg−1, was shown to reduce pulmonary oedema, lung vascular permeability, neutrophil infiltration and TNF-α and MIP-2 release, via an α7 cholinergic receptor-dependent mechanism in a model of acid aspiration-induced pneumonitis 8. In contrast to the model of acid aspiration-induced lung injury, which induces acute lung inflammation, experimentally induced pneumococcal pneumonia is associated with a gradual onset of inflammation and the development of an innate immune response over the first 24–48 h 13–15. Hence, rather than giving a single high dose, the current authors chose a nicotine dosing and treatment scheme that previously was demonstrated to reduce lethality and systemic inflammation induced by abdominal sepsis-caused CLP-induced faecal peritonitis (400 µg·kg−1 every 8 h) 7. This dose is in the same range as used in a model of influenza pneumonia, when nicotine, administered by mini-osmotic pumps at a rate of 1–2 mg·kg−1·day−1, reduced lung inflammation as measured by histopathology 10. Moreover, a nicotine dose within the same range was reported to be effective in reducing LPS-induced systemic inflammation 7, LPS-induced uveitis 22 and renal ischaemia/reperfusion injury 23. Together, these data indicate that nicotine, while effective in diminishing systemic and lung inflammation in several models, is not effective in reducing lung inflammation in the clinically relevant model of pneumococcal pneumonia. The apparent discrepancy between the current study and earlier studies on the effects of nicotine may not only be related to the differences in body compartments examined (lung versus abdominal cavity/circulation), but also to the type of causing micro-organism (Gram-positive versus Gram-negative). In this respect it would be interesting to study the effect of nicotine during Gram-negative pneumonia. It is of note that, at 24 h after infection, most parameters of inflammation (histopathology and cytokine concentrations) were exaggerated in mice treated with nicotine. This was most likely caused by the relatively increased bacterial loads (providing a more potent pro-inflammatory stimulus) at this time-point. Indeed, this “pro-inflammatory” effect of nicotine was only seen at the time-point where bacterial loads were higher (24 h) and not at either an earlier (4 h) or later (48 h) time-point; in addition, the extent of inflammation in this model closely follows the bacterial burden in the lungs 17, 24, and strong correlations were found between the pulmonary bacterial load and pro-inflammatory cytokine concentrations in the current study. This may also explain why at 24 h more neutrophils were present in the lungs of mice treated with nicotine. In this respect, it should be noted that neutrophils express several nicotinic receptors, including the α7 cholinergic receptor 25, and that stimulation of these receptors has been shown to inhibit rather than enhance neutrophil migration 26.
As mentioned, nicotine treatment was associated with higher bacterial loads at 24 h after infection. Considering that an adequate early inflammatory reaction in the lung is essential for mounting an effective host defence response in this model of pneumococcal pneumonia 17, it was speculated that nicotine might inhibit the innate immune response very early after infection and thereby facilitate subsequent bacterial growth. However, such an effect of nicotine could not be shown at 4 h after infection with S. pneumoniae (table 1⇑). An alternative explanation could be a relatively defective phagocytosis and/or killing of S. pneumoniae in the presence of nicotine. Indeed, nicotine has been reported to enhance the replication of Legionella pneumophila in cultures of mouse alveolar macrophages, which was associated with inhibition of the production of pro-inflammatory cytokines such as TNF-α and IL-12 27. Moreover, exposure of human neutrophils to nicotine reduced their ability to kill several oral bacterial pathogens 28. Ongoing current studies are examining in detail the effect of various cholinergic receptor agonists on antibacterial effector functions of different leukocyte subsets.
Although the present study did not examine the effect of inhaled nicotine during pneumococcal pneumonia, it is interesting to note that smokers and even nonsmokers exposed to second-hand smoke have an increased risk for invasive pneumococcal infection 29. A causative role for smoking in the pathogenesis of pneumococcal pneumonia is plausible, but has not been directly demonstrated. Clearly, smoking damages local immune defences within the airways and can enhance the binding of S. pneumoniae to pharyngeal cells 30. The current study has added to this that parenteral nicotine exposure transiently facilitates the growth of pneumococci in the lungs of mice experimentally infected with this pathogen.
In conclusion, the present study showed that nicotine, given at a dose and treatment schedule that was earlier found to protect mice from death due to cecal ligation and puncture-induced polymicrobial sepsis 7, transiently impairs antibacterial defence during pneumococcal pneumonia. Nicotine was unable to inhibit the inflammatory response to Streptococcus pneumoniae in the lung or the circulation: one day after infection, when bacterial loads were increased in nicotine-treated mice, pulmonary and systemic inflammation were even enhanced.
Statement of interest
None declared.
Acknowledgments
The authors would like to thank J. Daalhuisen and M. ten Brink (both Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands) for excellent technical assistance.
- Received July 7, 2008.
- Accepted September 19, 2008.
- © ERS Journals Ltd
References










