





Abstract
Pneumonia, the main cause of acute lung injury, is characterised by a local pro-inflammatory response and coagulopathy. Mechanical ventilation (MV) is often required. However, MV can lead to additional injury: so-called ventilator-induced lung injury (VILI). Therefore, the current authors investigated the effect of VILI on alveolar fibrin turnover in Streptococcus pneumoniae pneumonia.
Pneumonia was induced in rats, followed 48 h later by either lung-protective MV (lower tidal volumes (LVT) and positive end-expiratory pressure (PEEP)) or MV causing VILI (high tidal volumes (HVT) and zero end-expiratory pressure (ZEEP)) for 3 h. Nonventilated pneumonia rats and healthy rats served as controls. Thrombin–antithrombin complexes (TATc), as a measure for coagulation, and plasminogen activator activity, as a measure of fibrinolysis, were determined in bronchoalveolar lavage fluid (BALF) and serum.
Pneumonia was characterised by local (BALF) activation of coagulation, resulting in elevated TATc levels and attenuation of fibrinolysis compared with healthy controls. LVT-PEEP did not influence alveolar coagulation or fibrinolysis. HVT-ZEEP did intensify the local procoagulant response: TATc levels rose significantly and levels of the main inhibitor of fibrinolysis, plasminogen activator inhibitor-1, increased significantly. HVT-ZEEP also resulted in systemic elevation of TATc compared with LVT-PEEP.
Mechanical ventilation causing ventilator-induced lung injury increases pulmonary coagulopathy in an animal model of Streptococcus pneumoniae pneumonia and results in systemic coagulopathy.
Community-acquired pneumonia is the most frequent cause of acute lung injury (ALI) or its more severe form, acute respiratory distress syndrome (ARDS), often requiring mechanical ventilation (MV) 1. Although MV provides essential life support, it can also worsen lung injury: so-called ventilator-induced lung injury (VILI). One large multicentre trial established the importance of VILI by demonstrating that ventilation with lower tidal volumes (VT) versus traditional VT (6 versus 12 mL·kg−1) improves survival 2. The spectrum of VILI also encompasses increases in pulmonary and systemic inflammatory mediators known as biotrauma 3–6, which has been linked to multiple organ failure 7.
Pulmonary inflammation associated with pneumonia is characterised by local generation of pro-inflammatory mediators and a pro-coagulant shift of the alveolar haemostatic balance 8. The latter is the result of activation of coagulation on the one hand, which causes alveolar fibrin production, and attenuation of fibrin breakdown on the other 8, 9. Disturbances in alveolar fibrin turnover have been demonstrated in patients with pneumonia 10, 11 and ALI/ARDS 11.
Recently, it was demonstrated that in models of VILI, local pulmonary fibrinolysis is suppressed during injurious mechanical ventilation known as ventilator-associated coagulopathy 12, 13. However, the models used were iatrogenic in nature, either a single exposure to lipopolysaccharide 13 or fibrin deposits caused by human plasma without inflammation 12. Furthermore, in neither of these models was systemic analysis performed. It is unknown whether mechanical ventilation affects the pulmonary coagulopathy in pneumonia and if this can lead to systemic changes. In the current study, the effect of VILI on pulmonary and systemic coagulopathy in a clinically relevant model of Streptococcus pneumoniae pneumonia was determined.
MATERIALS AND METHODS
All studies were approved by the animal care committee at St Michael’s Hospital (Toronto, ON, Canada) in accordance with Canadian Council of Animal Care guidelines. A total of 57 male Sprague–Dawley rats (weight 240–300 g; Charles River, St Constant, QC, Canada) were included in the study.
Induction of pneumonia
As previously described 14, S. pneumoniae pneumonia was induced by intratracheal instillation of live bacteria in 51 rats. Briefly, ∼5×106 colony-forming units (CFU) of S. pneumoniae serotype 3 (ATCC 6303; ATCC, Manassas, VA, USA) per rat was aerosolised intratracheally using a trans-oral miniature nebulizer (Penn-Century, Philadelphia, PA, USA) 15 under light anaesthesia (65% nitrous oxide/33% oxygen/2% isoflurane). Animals were allowed to recover from anaesthesia and returned to their cages with food and water ad libitum. Rats were monitored every 8 h and received supplemental fluids every 24 h by i.p. injection (30 mL·kg−1 of lactated Ringer’s solution). A total of 15 nonventilated rats with pneumonia and six nonventilated rats without pneumonia served as controls (nonventilated pneumonia controls and healthy controls, respectively).
Experimental protocol
Animals were block randomised 48 h after intratracheal challenge with bacteria either to one of the two ventilation strategies or to no ventilation (fig. 1⇓). In total, 30 rats were anaesthetised with an i.p. injection of 100 mg·kg−1 ketamine (Ketalean 100 mg·mL−1; Bimeda-MTC Animal Health Inc., Cambridge, ON, Canada) and 6 mg·kg−1 xylazine (Rompun 20 mg·mL−1; Bayer Inc., Toronto). A sterile metal cannula was inserted into the trachea and a polyethylene catheter was inserted into the carotid artery. Anaesthesia was maintained by continuous i.v. infusion of ketamine (15 mg·kg−1·h−1) and xylazine (3 mg·kg−1·h−1). Paralysis was achieved by infusing pancuronium (0.35 mg·kg−1·h−1). Medications were infused via a tail-vein catheter. Animals were subsequently ventilated in the volume-controlled mode (Servo Ventilator 300; Maquet, Solna, Sweden), at an inspiratory oxygen fraction of 0.4 and an inspiration to expiration ratio of 1:2. VT was initially set at 6 mL·kg−1 body weight with a positive end-expiratory pressure (PEEP) of 0.5 kPa (5 cmH2O).

Schematic representation of the study design. LVT: lower tidal volume; PEEP: positive end-expiratory pressure; VT: tidal volume; HVT: higher VT; ZEEP: zero end-expiratory pressure. 1 cmH2O = 0.1 kPa.
After a 10-min stabilisation period, rats were randomised to either a lung-protective strategy using lower VT (LVT) and PEEP (n = 15) or a strategy causing VILI with higher VT (HVT; 12 mL·kg−1 body weight) and zero end-expiratory pressure (ZEEP; n = 15) as previously described 16. Settings were based on current knowledge that both increased VT and lack of PEEP are causative parts of VILI 7, 17, and aimed to keep mean airway pressures between the groups similar, hence minimising the effect on mean arterial blood pressure (MAP) 18. Animals were ventilated for 3 h. Blood pressure was continuously monitored through the intra-arterial catheter. Blood samples of 100 µL were taken hourly and cultured undiluted on sheep-blood agar plates, incubated and counted after 24 h. Blood gas determinations were performed hourly, using a pH/blood gas analyser (Ciba-Corning Model 248 blood gas analyser; Corning Medical, Medfield, MA, USA). Body temperature was maintained at 37°C using a heating pad. At the end of the experiment all animals were sacrificed by exsanguination.
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) was performed twice, with 30 mL·kg−1 body weight of saline, and pooled. The number of viable bacteria in the BAL fluid (BALF) was determined by plating 10-fold dilutions on sheep-blood agar plates: plates were incubated at 37°C in 5% CO2 and counted after 24 h. The BALF was centrifuged at 4°C at 400×g for 10 min, and the supernatant was snap-frozen on liquid nitrogen and stored at -80°C until further analysis.
Histology
For three animals from each group, lungs were taken for histopathology. Lungs were removed en bloc without BAL and fixed in 4% paraformaldehyde at a mean pressure of 1.0 kPa (10 cmH2O). The specimens were embedded in paraffin, sectioned in tissue blocks from all lobes and stained with haematoxylin and eosin. The analysing pathologist was not informed of the study purpose, was blinded to the sample identity and only asked to score for lung injury. Scoring categories of none, mild, moderate and severe (scores 0, 1, 2 and 3, respectively) were assigned for descriptive purposes as previously described 19, for each of the following criteria: alveolar collapse; alveolar haemorrhage; perivascular oedema; polymorphonuclear leukocyte infiltration; alveolar membranes; and alveolar oedema.
Coagulation and fibrinolysis
Levels of thrombin-antithrombin complexes (TATc), plasminogen activator inhibitor (PAI)-1 and D-dimers were measured by ELISA in BALF supernatants in accordance with the manufacturer’s instructions (TATc: Behring, Marburg, Germany; PAI-1: Biopool, Umeå, Sweden; Asserachrom D-dimer: Diagnostica Stago, Asnières-sur-Seine, France). Plasminogen activator activity (PAA) and levels of antithrombin (AT) were measured using an amidolytical assay 20. Measurements were performed simultaneously in BALF and plasma.
Cytokines
Concentrations of tumour necrosis factor (TNF)-α and interleukin (IL)-6 in BALF supernatants and plasma were measured using rat-specific ELISA kits (BioSource, Camarillo, CA, USA) in accordance with the manufacturer’s instructions.
Statistical analysis
Since the primary objective of the present study was the effect of different ventilation strategies on pneumonia, only the three pneumonia groups were compared (HVT-ZEEP, LVT-PEEP and nonventilated pneumonia group). Intergroup differences were analysed by ANOVA. If ANOVA resulted in p-values <0.05, a Bonferroni post hoc test was performed. A value of p<0.05 was considered statistically significant. All data are presented as mean±sem.
RESULTS
Pneumonia
After intratracheal challenge with S. pneumoniae, all animals developed severe pneumonia, resulting in the death of six (12%) of the infected animals. The severity of the pneumonia was also evident by both macroscopic (a minimum of 25% of the lungs was visually affected at autopsy) and microscopic histological examinations.
Microbiological examinations
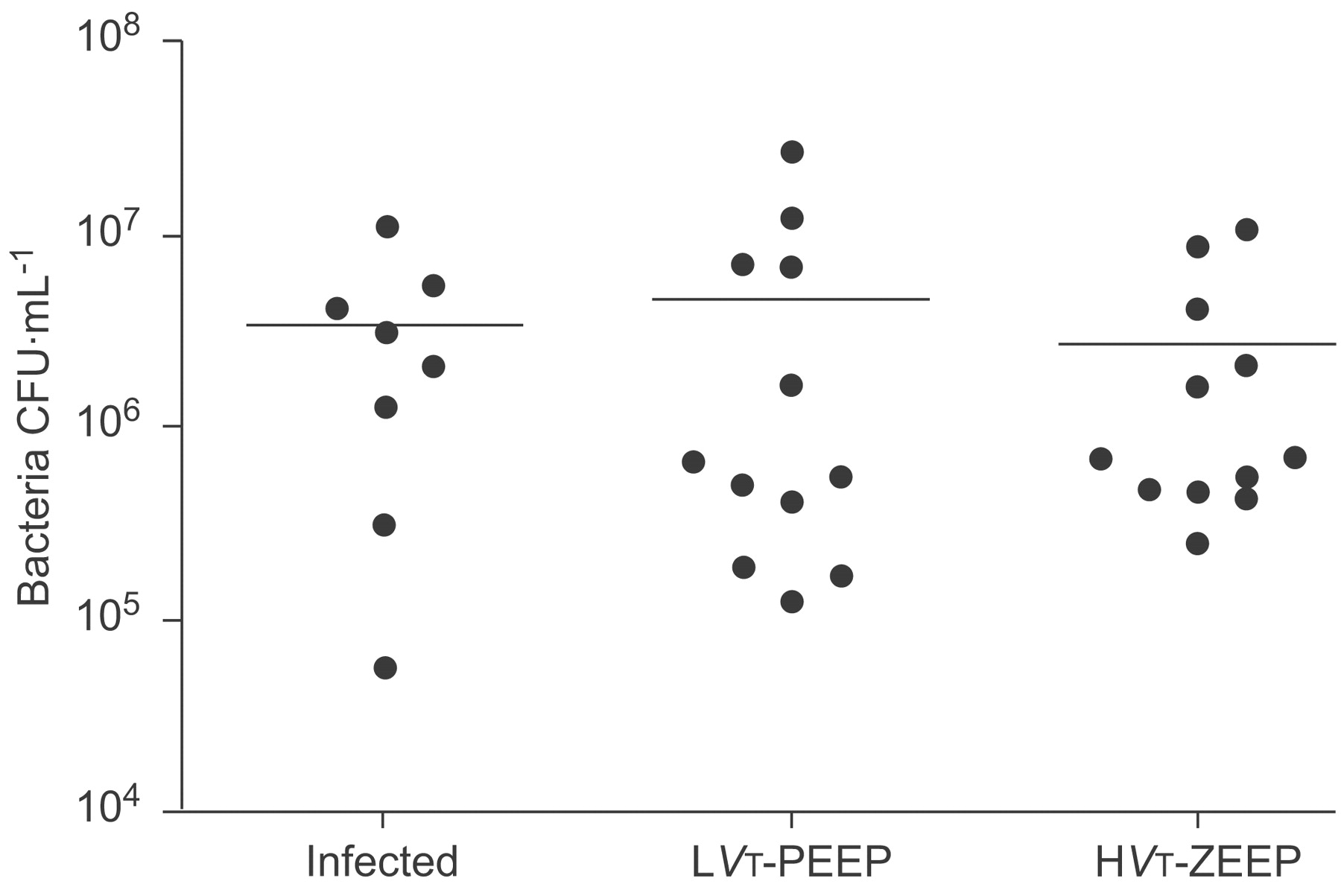
There was no difference in the number of bacteria in BALF between nonventilated pneumonia rats and mechanically ventilated rats (fig. 2⇓). Also, there was no difference in the number of bacteria in BALF between animals ventilated with the LVT-PEEP strategy and animals subjected to HVT-ZEEP. Similarly, the number of animals with bacteraemia did not differ significantly: eight out of 15 rats ventilated with the LVT-PEEP strategy, and seven out of 15 rats with HVT-ZEEP showed positive blood cultures.

Number of bacterial colony-forming units (CFU) in bronchoalveolar lavage fluid at the end of the experiment in the three groups with pneumonia. Infected: control group with Streptococcus pneumoniae pneumonia, not ventilated; lower tidal volume (LVT) and positive end-expiratory pressure (PEEP): group with pneumonia and ventilated with lung-protective mechanical ventilation; higher tidal volume (HVT) and zero end-expiratory pressure (ZEEP): group with pneumonia and mechanical ventilation causing ventilator-induced lung injury.
Gas exchange and MAP
There were no differences in MAP during the period of MV (fig. 3a⇓). There were also no differences in oxygenation in either MV group over time (fig. 3b⇓). All ventilated animals with pneumonia required i.v. fluid administration at the start of MV because of hypotension, but no differences existed between the two MV groups (25±5 and 24±5 mL for HVT-ZEEP and LVT-PEEP, respectively).

a) Mean arterial blood pressure (MAP) over time in the two ventilated pneumonia groups. b) Mean arterial oxygenation over time in the two ventilated pneumonia groups, measured as arterial oxygen tension (Pa,O2) divided by inspiratory oxygen fraction (FI,O2). ○: group with pneumonia and ventilated with lung-protective mechanical ventilation; •: group with pneumonia and mechanical ventilation causing ventilator-induced lung injury. 1 mmHg = 0.133 kPa.
Pulmonary coagulation
S. pneumoniae pneumonia caused local activation of coagulation: BALF levels of TATc increased from 0.9±0.2 ng·mL−1 in healthy rats to 4.0±0.5 ng·mL−1 in nonventilated pneumonia rats (fig. 4⇓). While LVT-PEEP did not alter BALF levels of TATc (4.0±0.5 ng·mL−1), HVT-ZEEP resulted in a significant increase of BALF levels of TATc to 5.5±0.7 ng·mL−1 (p<0.001 versus LVT-PEEP). BALF levels of the endogenous anticoagulant AT were highest in the healthy animals (20.0±4.7 IU·mL−1) and were depressed in all pneumonia groups. Although BALF levels of AT were lowest in the HVT-ZEEP group, differences with LVT-PEEP rats did not reach statistical significance.

Effect of pneumonia alone and mechanical ventilation and pneumonia on a) thrombin-antithrombin complex (TATc), b) antithrombin (AT), c) plasminogen activator activity (PAA), d) plasminogen activator inhibitor (PAI)-1 levels and e) D-dimers in bronchoalveolar lavage fluid (BALF). Noninfected: control group without pneumonia and not ventilated; infected: control group with Streptococcus pneumoniae pneumonia, not ventilated; lower tidal volume (LVT) and positive end-expiratory pressure (PEEP): group with pneumonia and ventilated with lung-protective mechanical ventilation; higher tidal volume (HVT) and zero end-expiratory pressure (ZEEP): group with pneumonia and mechanical ventilation causing ventilator-induced lung injury. *: p<0.05 for comparison of the infected control and LVT-PEEP groups with the HVT-ZEEP group.
Pulmonary fibrinolysis
PAA levels in BALF were highest in healthy rats (99.5±7.0%); pneumonia led to a decrease (71.5±10.3%; fig. 4⇑). LVT-PEEP did not alter PAA levels in BALF (71.4±7.0%), while injurious ventilation further decreased PAA levels (61.4±7.5%). BALF levels of D-dimers were increased in all rats with pneumonia compared with healthy controls (296±44 versus 13±3 ng·mL−1, respectively). There was no effect of either MV strategy on BALF levels of D-dimers. BALF levels of PAI-1, the main inhibitor of fibrinolysis, were higher in nonventilated pneumonia rats compared with healthy animals (11.1±1.7 versus 1.4±0.5 ng·mL−1, respectively). While LVT-PEEP did not alter PAI-1 levels in BALF (11.6±1.7 ng·mL−1), HVT-ZEEP significantly increased PAI-1 levels (15.5±1.7 ng·mL−1; p<0.001 versus LVT-PEEP).
Systemic coagulation and fibrinolysis
Activation of coagulation was also observed in plasma of nonventilated pneumonia controls compared with healthy rats (TATc levels 12.7±1.0 versus 4.7±0.7 ng·mL−1, respectively; fig. 5⇓). HVT-ZEEP increased TATc in plasma (15.3±2.0 ng·mL−1; p<0.05 versus LVT-PEEP and infected controls), while LVT-PEEP did not alter systemic TATc levels (12.5±1.8 ng·mL−1). Systemic PAA levels were significantly higher in healthy control animals compared with nonventilated pneumonia rats (p<0.05), but this was not altered by the MV strategies.

Effect of pneumonia alone and mechanical ventilation and pneumonia on a) thrombin-antithrombin complex (TATc) and b) plasminogen activator activity (PAA) levels in plasma. Noninfected: control group without pneumonia and not ventilated; infected: control group with Streptococcus pneumoniae pneumonia, not ventilated; lower tidal volume (LVT) and positive end-expiratory pressure (PEEP): group with pneumonia and ventilated with lung-protective mechanical ventilation; higher tidal volume (HVT) and zero end-expiratory pressure (ZEEP): group with pneumonia and mechanical ventilation causing ventilator-induced lung injury. *: p<0.05 for comparison of the infected control and LVT-PEEP groups with the HVT-ZEEP group.
Cytokines
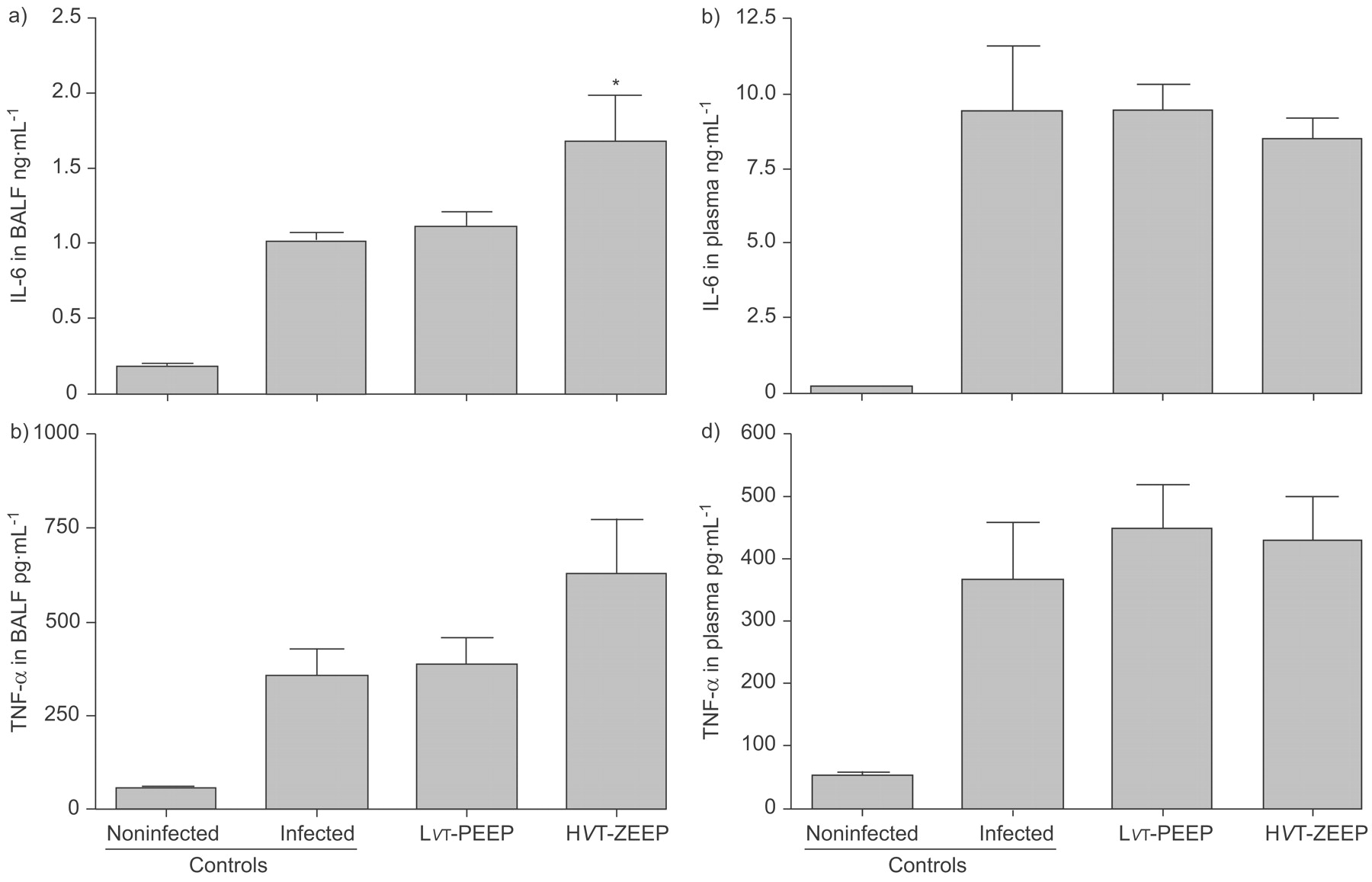
IL-6 levels were increased both in BALF and plasma of nonventilated pneumonia controls compared with healthy rats (BALF: 1.03±0.05 versus 0.19±0.01 ng·mL−1, respectively; plasma: 9.4±2.2 versus 0.15±0.01 ng·mL−1, respectively; fig. 6⇓). HVT-ZEEP further increased IL-6 levels in BALF (1.69±0.31 ng·mL−1; p<0.05 versus infected controls), while LVT-PEEP did not alter BALF IL-6 levels. Ventilation did not affect systemic IL-6 levels.

Effect of pneumonia alone and mechanical ventilation and pneumonia on a) interleukin (IL)-6 levels in bronchoalveolar lavage fluid (BALF) and b) plasma, and on c) tumour necrosis factor (TNF)-α levels in BALF and d) plasma. Noninfected: control group without pneumonia and not ventilated; infected: control group with Streptococcus pneumoniae pneumonia, not ventilated; lower tidal volume (LVT) and positive end-expiratory pressure (PEEP): group with pneumonia and ventilated with lung-protective mechanical ventilation; higher tidal volume (HVT) and zero end-expiratory pressure (ZEEP): group with pneumonia and mechanical ventilation causing ventilator-induced lung injury. *: p<0.05 for comparison of the infected control and LVT-PEEP groups with the HVT-ZEEP group.
TNF-α was increased in BALF of nonventilated pneumonia controls compared with healthy rats (355±72 versus 51±4 pg·mL−1, respectively; fig. 6⇑). LVT-PEEP did not change TNF-α levels (388±68 pg·mL−1) and HVT-ZEEP resulted in the highest levels (632±141 pg·mL−1), although this was not significantly different from other groups. Systemic TNF-α levels were higher in nonventilated pneumonia rats compared with healthy control animals (367±90 versus 52±5 pg·mL−1, respectively), but this was not altered by the MV strategies.
Histopathology
Histological analysis clearly showed pneumonia in all infected animals (fig. 7⇓). HVT-ZEEP resulted in increased infiltration of neutrophils and alveolar collapse, which was significantly higher compared with LVT-PEEP (n = 3 in each group).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative sections of lungs stained with haematoxolin and eosin. a and d) Infected: control group with Streptococcus pneumoniae pneumonia, not ventilated. b and e) Lower tidal volume (LVT) and positive end-expiratory pressure (PEEP): group with pneumonia and ventilated with lung-protective mechanical ventilation. c and f) Higher tidal volume (HVT) and zero end-expiratory pressure (ZEEP): group with pneumonia and mechanical ventilation causing ventilator-induced lung injury. g) Lung injury score data, shown as mean±sem. *: p<0.05 for HVT-ZEEP versus LVT-PEEP. Scale bars = 100 μm.
DISCUSSION
The main finding of the present study was that in lungs infected with S. pneumoniae, HVT-ZEEP promoted activation of coagulation and attenuation of fibrinolysis, while LVT-PEEP left the procoagulant state in pneumonia unaltered. As far as the current authors know, the present study is among the first to show that MV can aggravate pulmonary coagulopathy in pneumonia. Importantly, these haemostatic changes were also found in the systemic compartment. Notably, differences in coagulation activation and fibrinolysis attenuation could not be explained by differences in numbers of bacteria present in the lung. Indeed, the bacterial burden was not different after 3 h of MV in the two MV groups. In addition, no differences in oxygenation and systemic haemodynamics were found during the course of MV.
In patients with established community-acquired or ventilator-associated pneumonia 11, 20, and in intubated and mechanically ventilated patients developing pulmonary infection 8, 10, there are procoagulant changes in the pulmonary compartment. Concomitant depressed levels of endogenous anticoagulant proteins and depressed fibrinolytic activity is also observed, and the combination of these changes leads to alveolar fibrin depositions. The findings in the present rat model of S. pneumoniae pneumonia closely resemble those found in clinical studies. Similar to patients with pneumonia, coagulation was activated and fibrinolysis was attenuated in the pulmonary compartment of animals after challenge with bacteria. Also, levels of AT were lower after induction of pneumonia. Therefore, the model can be considered of interest for preclinical studies on ventilator-induced coagulopathy, at least in the setting of pneumonia. Although animal models will never be able to simulate the complex diseases in patients and the time of ventilation is also shorter than those in patients (3 h versus days), the observation that even a short period of injurious ventilation in pneumonia causes coagulopathy emphasises the consequences of injurious ventilation in general and specifically in high-risk groups such as pneumonia. It remains speculative whether the present model has similarities with ALI/ARDS, although the findings in the pulmonary compartment in patients with ALI/ARDS closely resemble those found in patients with pneumonia 11.
S. pneumoniae was specifically chosen as the causative pathogen in the current experimental setup because community-acquired pneumonia is most often caused by this pathogen, which is responsible for >500,000 cases of lower respiratory tract infection in the USA each year 21. Ventilator-associated pneumonia is commonly caused by Pseudomonas species and other Gram-negative bacteria 22. Similar changes in alveolar fibrin turnover, however, have been found in patients suffering from ventilator-associated pneumonia 10, 11. Furthermore, in models of P. aeruginosa pneumonia, comparable procoagulant changes in BALF have been described 23. Importantly, in patients with ventilator-associated pneumonia, it was recently shown that nonsurvivors showed significantly higher BALF levels of TATc and PAI-1 than survivors 24, and increased PAI-1 levels also correlated with mortality and adverse clinical outcome in patients from the ARDS-network study 25. Similarly, higher systemic PAI-1 levels are associated with poor prognosis in septic patients 26. Although the direct link between TATc and PAI-1 levels and mortality needs to be proven first, these data are very suggestive that MV strategies that promote coagulation and attenuate fibrinolysis may worsen prognosis.
MV alone has been demonstrated to affect pulmonary coagulation in other preclinical animal models, as well as in one clinical study 12, 13, 27. It was recently shown in a rat model of MV that the use of increasing VT, but similar PEEP levels, caused a shift in alveolar fibrin turnover, mainly due to higher levels of PAI-1 in the lungs of these animals 12, 13. In addition, a recent study reported on patients without pre-existing lung injury who were intubated and mechanically ventilated because of elective surgery 27. In these patients, 5 h of MV with traditional VT (12 mL·kg−1 ideal body weight) and no PEEP caused activation of pulmonary coagulation as reflected by a marked increase in TATc levels in BALF, while lung-protective MV (using VT of 6 mL·kg−1 predicted body weight and 1.0 kPa (10 cmH2O) PEEP) did not cause a rise in TATc levels 27. Based on these findings the current authors suggest that coagulopathy may be an intrinsic component of VILI (or its clinical counterpart, ventilator-associated lung injury).
Insights into the pathophysiology of VILI come from animal studies that demonstrated that MV with larger VT rapidly results in pulmonary changes that mimic ARDS 28. Injurious MV settings resulted in recruitment and activation of inflammatory cells 29, local production of inflammatory mediators (e.g. cytokines) 30 and leakage of such mediators into the systemic circulation 4. Ranieri and co-workers 5, 31 confirmed a reduction in BALF and systemic concentrations of inflammatory mediators during lung-protective MV compared with conventional MV in a clinical trial. In the present study, IL-6 and TNF-α were both increased in all the pneumonia animals; however, only VILI increased IL-6 in the lung. TNF-α, an early-release cytokine with a peak response at 1–2 h, was not increased, which corroborates previous findings 32. In a recent study using gene profiling, IL-6 was shown to be one of the key cytokines to be affected during VILI while no effect was seen on TNF-α 33, which is similar to the observations of the present study.
Although many studies have shown impairment of oxygenation to be a hallmark of lung injury, in the present model, gas exchange was not impaired, which is similar to the results of Verbrugge et al. 34. The oxygenation also did not change during the period of ventilation, most likely due to the VT used in the current model (12 mL·kg−1), which is considerably lower than other animal studies (VT ≤45 mL·kg−1). Additionally, improved gas exchange with the higher VT of 12 mL·kg−1 did not improve outcome in patients with ALI 2.
The current study has several limitations, such as the fact that animals with S. pneumoniae pneumonia were ventilated; thus, only the additional effect of ventilation on pre-existing lung injury (coagulopathy) in a single model of pulmonary ALI can be demonstrated. Furthermore, the applied ventilator strategies cannot separate the individual effects of either VT or PEEP on the observed ventilator-induced coagulopathy. These limitations should be addressed in future studies.
In conclusion, injurious ventilation settings promote procoagulant changes and attenuate fibrinolysis in a rat model of pneumonia. These changes are not restricted to the site of inflammation, since changes in fibrin turnover were also found in the systemic compartment with injurious ventilation settings.
Support statement
J.J. Haitsma is a recipient of the Eli Lilly–University of Toronto, Critical Care Medicine Fellowship. M.J. Schultz is supported by a personal grant from the Netherlands Organization for Health Research and Development (ZonMW), NWO-VENI grant 2004 (project number 016.056.001). J.W. Kuiper is supported by a stipend from the Ter Meulen Fund, Royal Netherlands Academy of Arts and Sciences, the Netherlands. H. Zhang and A.S. Slutsky are supported by the Canadian Institutes of Health Research (Ottawa, ON, Canada).
Statement of interest
None declared.
- Received March 25, 2008.
- Accepted August 18, 2008.
- © ERS Journals Ltd
References










