





Abstract
Chronic obstructive pulmonary disease is a leading global cause of morbidity and mortality that is characterised by inexorable deterioration of small airways obstruction with emphysema associated with cellular inflammation and structural remodelling. Other features include apoptosis as well as proliferation of cells, and both tissue repair and lack of tissue repair.
Metalloprotease release, together with that of apoptotic factors, may underlie the emphysema, and, conversely, fibrosis of the small airways may be accounted for by the effects of growth factor activation. In advanced disease, influential factors include the development of autoimmunity, with activation of dendritic cells and T-helper cells of both type 1 and 2, and the senescence response.
An inability of macrophages to ingest apoptosed cells and bacteria may exacerbate inflammatory responses. Systemic inflammation with concomitant cardiovascular disease and metabolic syndrome may reflect the effect of cigarette smoke on nonpulmonary cells. Corticosteroid resistance may be secondary to oxidative stress mechanisms, such as inactivation of histone deacetylases.
The mechanisms of chronic obstructive pulmonary disease may be heterogeneous, according to severity, and clinical phenotypes need to be correlated with cellular and pathological processes. Treatments may be targeted to patients with specific mechanisms.
SERIES “CELL AND ANIMAL STUDIES IN RESPIRATORY MEDICINE”
Edited by R. Farré and A.T. Dinh-Xuan
Number 2 in this Series
Chronic obstructive pulmonary disease (COPD) is a leading global cause of morbidity and mortality, and will continue to increase in importance as the world population continues to age 1. The recent Burden of Obstructive Lung Disease initiative study on the global prevalence of Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage II COPD, defined as being of moderate severity, showed worldwide prevalences of 5.1–16.7% in females and 8.5–22.2% in males 2. The pooled global prevalence in adults aged >40 yrs is estimated to be 9–10%. The estimated death rate in various countries is variable, with the reported mortality ranging from 4.4 per 100,000 population in Japan to 130 per 100,000 population in China. The prediction is that COPD will become the fifth most frequent burden of disease worldwide 3. Deaths may be caused not only by respiratory causes, usually respiratory failure, but also by lung cancer, cardiovascular disease and other causes, often unidentifiable 4. Admission to hospitals due to COPD contributes most to the direct medical costs of COPD in many high-income countries 5.
COPD is defined as a preventable and treatable disease with some significant extrapulmonary effects that may contribute to severity in individual patients 6. Its pulmonary component is characterised by airflow limitation that is not fully reversible but usually progressive and associated with an abnormal inflammatory response of the lung to noxious particles or gases. Although cigarette smoking is the main pathological driver of COPD, other factors may be involved, including a genetic predisposition that could explain why only a proportion of cigarette smokers develop COPD, other particulates or gases in environmental pollution or exposure to biomass combustion (to explain why some patients who develop COPD are never-smokers), bacterial or viral infections (acting as amplifiers of the established disease), and bronchial hyperresponsiveness (with the Dutch hypothesis that this abnormality predisposes to both COPD and asthma).
Pathologically, distinct disease processes are recognised: chronic bronchitis, emphysema and small airways disease. These affect all parts of the lungs, including large and small airways and parenchyma, and contribute to the chronic airflow obstruction (as measured by a reduction in forced expiratory volume in one second (FEV1) or the ratio of FEV1 to forced vital capacity) through increases in the resistance of the conducting airways and lung compliance. Chronic bronchitis is a clinical description for a chronic increase in bronchial secretions, characterised by a productive cough with a pathological correlate of submucosal glandular hypertrophy and hyperplasia, with dilated ducts in airways down to 2–4 mm in internal diameter. Emphysema is an anatomicopathological diagnosis defined by permanent destructive enlargement of airspaces distal to the terminal bronchioles, contributing to airflow limitation resulting from loss of lung elastic recoil 7. Chronic inflammation and remodelling of the small airways and particularly of the terminal bronchioles (epithelial metaplasia, increased airway smooth muscle and goblet cell hyperplasia and submucosal gland hypertrophy) are features related to the severity of disease characteristic of COPD 8. The degree of airflow limitation, as measured by FEV1, is also correlated with the degree of airway wall thickness, providing indirect evidence for a role for airway wall remodelling in airflow obstruction in COPD.
At the cellular level (fig. 1⇓), many biological processes characterise the development of COPD. Cigarette smokers develop some degree of lung inflammation, but the COPD patient develops a far greater degree of inflammation that progresses rapidly with advanced disease, often accompanied by systemic inflammation and inflammation in the heart, blood vessels and skeletal muscle. Squamous cell metaplasia and cell atypia are features of cigarette smokers, and these changes may be precursors of cancer development; conversely, apoptosis is one potential mechanism of alveolar destruction. Breakdown of extracellular matrix occurs in parenchymal tissues, with a simultaneous increase in extracellular matrix production in the adjacent small airways, as determined by the micro-environment. Oxidative stress may lead to the activation of many intracellular pathways, including kinases, transcription factors and epigenetic events that modulate the inflammatory response and cell cycling.
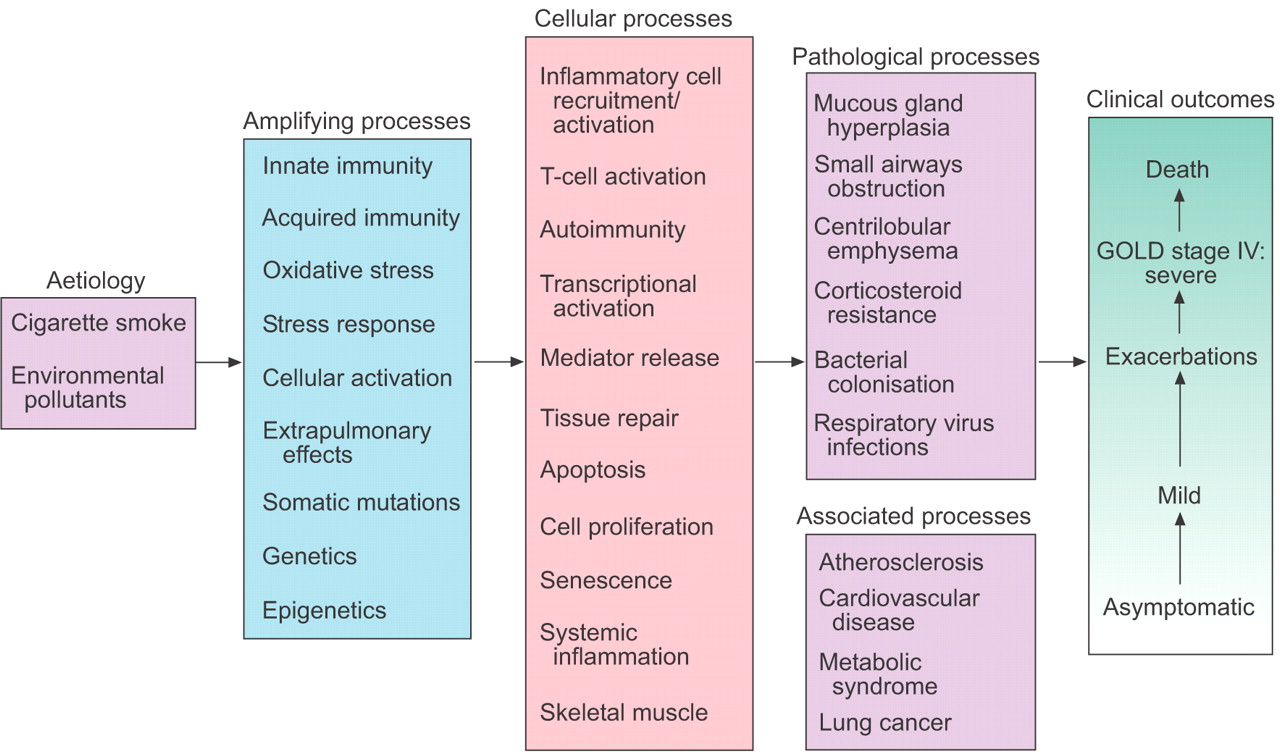
Amplification, cellular and pathological processes that link the aetiology of chronic obstructive pulmonary disease to clinical outcomes. GOLD: Global Initiative for Chronic Obstructive Lung Disease.
STUDYING MECHANISMS OF COPD
There have been several reviews of the pathophysiology of COPD 9–14. The current paper provides an overview of the important abnormal cellular and molecular processes that underlie the pathophysiology of COPD, examining to what extent studies of these processes have led to the discovery of new targets for drugs for the treatment of COPD. It particularly focuses on the recent literature that provides new insights and mechanisms of COPD, emphasising the multifaceted intrigue of lung inflammation, innate and acquired immunity, and tissue destruction and repair. Novel observations on the disease, which arise from studying the lungs and airways of patients, are purely descriptive but provide an array of hypotheses for testing in animal or cellular models. Since cigarette smoke is one of the main causes of COPD, the main thrust of research in COPD has been the study of the effect of cigarette smoke in whole animals or on cells in vitro, or the study of cells or tissues from patients with COPD. Moving from the patient to the animal or cellular model and back represents the iterative process by which an understanding of the complex pathophysiology of COPD can be unravelled.
In the analysis of tissues and cells obtained from patients with COPD, many studies include a group of healthy asymptomatic smokers, usually of the same age group and similar tobacco exposure as the group of patients with COPD, although often disease status in COPD may only be ascertained by a detailed examination of lung function and lung structure (e.g. using computed tomographic analysis of the lungs and airways). In addition, there is varying severity of disease cross-sectionally, i.e. graded according to spirometric performance and symptoms, as well as heterogeneity of disease (e.g. emphysema versus bronchial inflammation) that can make cohorts of COPD patients differ.
Another caveat in the analysis of studies on COPD is the degree of exposure to the offending stimulus, usually cigarette smoking, which is a complex mixture of gases, particles and up to 4,700 chemicals in a volatile phase, that could result in intersubject exposure variation in terms of cigarette brand, pattern of smoking and extent of exposure to other environmental factors, such as environmental pollution. Such differences must be taken into consideration when analysing the effect of cigarette smoke to which animals are exposed, since exposure protocols (secondary exposure) for these animals remain to be standardised as to the type and number of cigarettes, duration of exposure, etc. In isolated cells, exposure is performed using soluble extracts of cigarette smoke, often free of particulates, and may, therefore, not mimic the situation in vivo. It is also worth remembering that cigarette smoke contains 1×1015 free radical molecules in the gas phase and 1×1018 free radical molecules in the tar phase, and includes reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), hydroxyl anions and organic radicals. Therefore, cigarette smoke, through the induction of oxidative stress, not only causes COPD but also induces changes in circulation, cancerous changes in the epithelium and atherosclerosis, which may all contribute to the systemic component of inflammation that is an important aspect of the disease.
AIRWAY INFLAMMATION IN COPD
The chronic inflammation of COPD is characterised by an accumulation of neutrophils, macrophages, B-cells, lymphoid aggregates and CD8+ T-cells, particularly in the small airways 15, and the degree of inflammation increases with the severity of disease as classified by the GOLD (fig. 2⇓) 8.

Summary of inflammatory and cellular interactions linking chronic cigarette exposure to the chronic inflammation of chronic obstructive pulmonary disease (COPD). Activation of neutrophils, macrophages, epithelial cells, dendritic cells, T-cells, B-cells, fibroblasts and airway smooth muscle cells leads to release of cytokines, chemokines and proteases. Amplification signals are important in augmenting the inflammatory responses that underpin COPD. Ab: antibody; Th: T-helper cell; MHC: major histocompatibility complex; TCR: T-cell receptor; CXCL: CXC chemokine ligand; IP: interferon (IFN)-γ-inducible protein; CCL: CC chemokine ligand; RANTES: regulated on activation, normal T-cell expressed and secreted; TSLP: thymic stromal lymphopoietin; IL: interleukin; TNF: tumour necrosis factor; MCP: monocyte chemotactic protein; LT: leukotriene; CRP: C-reactive protein; TGF: transforming growth factor; EGF: epidermal growth factor; VEGF: vascular endothelial growth factor; MMP: matrix metalloproteinase.
Neutrophils
Neutrophils are able to release oxygen radicals, elastase and cytokines that are essential to the pathogenesis of COPD, with effects on goblet cells and submucosal glands, and in the induction of emphysema and inflammation. Neutrophils are localised particularly to the bronchial epithelium and bronchial glands 16 and also in close apposition to airway smooth muscle bundles 17; they are particularly found in the airway lumen, as recovered in sputum or by bronchoalveolar lavage (BAL). Sputum neutrophilia is increased in advanced COPD and is associated with the presence of greater airflow obstruction, particularly peripheral airflow obstruction, together with an accelerated decline in lung function 18–20. Sputum and circulating neutrophils from COPD patients express more of the leukocyte-specific integrin CD11b/CD18 21, 22, but blood neutrophils show impaired chemotaxis and migration to N-formyl-methionyl-leucyl-phenylalanine and interleukin (IL)-8 23. Cigarette smoke extract impairs the phagocytic ability of neutrophils, through suppression of caspase-3-like activity in the neutrophils, an impairment that does not lead to a suppression of spontaneous apoptosis 24. These abnormalities may underlie the increased risk of respiratory infections in smokers and COPD patients.
Eosinophils
Increased numbers of eosinophils have been reported in sputum, BAL fluid and the airway wall, with increased levels of eosinophil cationic protein in BAL fluid and induced sputum 25–29. Expression of IL-4 and -5, which are T-helper cell (Th) type 2-derived cytokines associated with eosinophilia of asthma, has been reported in plasma cells associated with submucosal glands 30. IL-8, in addition to its neutrophil chemotactic effects, also has eosinophil chemotactic properties 31. During exacerbations of chronic bronchitis, a marked increase in expression of RANTES (regulated on activation, normal T-cell expressed and secreted) has been reported in epithelium and subepithelium associated with a marked increase in submucosal eosinophil numbers 32. Expression of IL-5 is also increased 33. The role of eosinophils in the pathogenesis of COPD is unclear but they may represent a distinct subgroup of COPD. Increased numbers of eosinophils in sputum and BAL fluid in COPD have been related to a good clinical response to corticosteroid treatment 27, 34, 35. A recent study reported that a preferential distribution of eosinophils towards the airway lumen (i.e. low eosinophil numbers on biopsy with a high percentage of eosinophils in sputum) characterised patients with COPD with symptoms of chronic bronchitis compared with those without these symptoms 36.
Mast cells
Increased numbers of mast cells have been reported in COPD airways 37, 38, and in chronic bronchitis without airflow obstruction compared with airflow obstruction 30; other studies have not reported such findings 39, 40.
T-cells
CD8+ T-cell numbers are increased throughout the airways and in lung parenchyma 41 and have also been localised to airway smooth muscle bundles 17. Despite their prominence in COPD airways, their role still remains speculative. A study of CD8+ T-cell-deficient mice provides support for a role for these cells in the inflammatory response and emphysema development following long-term exposure to cigarette smoke through the production of the interferon (IFN)-γ-inducible CXC chemokine ligand (CXCL) 10 42. Lung parenchymal cells may be damaged by the release of lytic substances, such as perforin and granzyme, from CD8+ T-cells, and studies of COPD CD8+ T-cells reveal increased cytotoxic activity of these cells with higher concentrations of perforin present in sputum 43. In BAL fluid, soluble granzyme B levels and the percentage of T-cells expressing intracellular granzyme B and/or perforin were increased in both COPD groups and asymptomatic smokers, whereas soluble granzyme B levels were undetectable in the BAL fluid of nonsmokers 44. There is an association between apoptosing cell numbers and CD8+ T-cell numbers in alveolar walls 45, indicating a possible induction of apoptosis of epithelial and endothelial cells by CD8+ T-cells. One of the functions of CD8+ T-cells is to get rid of virally infected cells by cytolysis or apoptosis of such cells 46.
Activated CD4+ T-cell numbers are also increased in the small airway wall of smokers with severe COPD 47, and they appear to be of Th1 type, with expression of the chemokine CXCL10, which may control the release of elastolytic matrix metalloproteinases (MMPs) 48. In murine models, emphysema has been shown to develop in the presence of adoptively transferred pathogenic CD4+ T-cells. In severe emphysema, T-cells isolated from lung tissues showed oligoclonal expansion to conventional antigenic stimuli 49. Anti-elastin antibody, an autoantigen that may underlie the autoimmune response, has been reported in COPD patients 50. In addition, significantly fewer regulatory T-cells were found in the lungs of patients with COPD, with reduced gene expression of forkhead box protein (FOX) P3, a transcription factor crucial for the development of regulatory T-cells, and less IL-10 secretion, which could permit clonal expansion of elastin-specific Th1 cells 50. In contrast, in cells recovered by BAL, increased CD4/CD25 expression was reported in smokers and COPD patients compared with nonsmokers, with increased FOXP3 expression in these cells 51. The reason for this discrepant result, compared with those reported in lung tissue, remains unclear. Invariant killer T-cell numbers are not increased in COPD as assessed in sputum, BAL fluid and biopsy specimens from central airways 52.
Dendritic cells
The observation 8, 53 of increased numbers of B-cells and the presence of bronchus-associated lymphoid tissue in advanced COPD (not usually present in healthy nonsmokers) may reflect an adaptive immune response to chronic infection that is frequent at this stage; B-cells may also be the key link between the innate and adaptive immune responses. Lymphoid follicles consisting of B-cells and follicular dendritic cells (DCs) with adjacent T-cells were demonstrated in both the parenchyma and bronchial walls of patients with emphysema 54, and an oligoclonal antigen-specific reaction of the B-cells has been described. Plasma cells, which are derived from maturation of B-cells, were found to occur in greater numbers in subepithelial and submucosal glands in COPD compared with asymptomatic smokers, and a majority of these cells expressed IL-4 and -5 30. The increase in B-cell number may also reflect a role for autoimmune responses as a source of autoantibodies, and this is also supported by the reduced numbers of T-regulatory cells reported in lungs of COPD patients 50.
The idea of autoimmunity as a mechanism for the chronic inflammatory and emphysematous damage 55, 56 is also supported by the detection of circulating autoantibodies directed against proteins that could have been damaged following chronic cigarette smoke exposure, such as anti-elastin antibodies 50, anti-epithelial antibodies 57 and tobacco anti-idiotypic antibodies 58, in smoking patients with COPD. The anti-epithelial antibodies were particularly avid against pulmonary epithelium and endothelium, and mediated antibody-directed cytotoxicity, making them potentially pathogenic. Further support for the autoimmune pathogenesis comes from the development of an emphysema model in mice immunised with endothelial cells 46.
The accumulation of DCs detected as Langerhans’ cells in the epithelium and adventitia of small airways of patients with COPD with increasing severity 59, 60 supports the involvement of adaptive immunity; such an increase correlated with sputum levels of CC chemokine ligand (CCL) 20/macrophage inflammatory protein (MIP)-3α, a chemoattractant for DCs. However, another study found that bronchial mucosal DC numbers detected by ultrastructural morphology were reduced in current COPD smokers compared with ex-smokers with COPD, with DC numbers being similar to those in nonsmoking asthmatics and nonsmoking healthy controls 61. Whether these conflicting data might be due to assessing DCs at different stages of maturity is unclear. Further evidence for recruitment of DCs by cigarette smoke exposure comes from studies in mice in which chronic cigarette exposure led to an increase in CD11c+ DCs, associated with increased levels of CCL-2/monocyte chemotactic protein (MCP)-1 and CCL20/MIP-3α 62. Mice deficient in CC chemokine receptor (CCR) 6, expressed on pulmonary DCs and B-cells, showed less emphysema following cigarette smoke exposure 63, possibly through the inhibition of MMP-12 release from DCs 64. Other mechanisms for the initiation and perpetuation of airway inflammation and emphysema by DCs following cigarette smoke exposure remain to be studied.
Another mechanism by which DCs could be involved in COPD is through inhibition of Th1 immunity and preferential induction of Th2 responses. Cigarette smoke extract inhibited the DC-mediated priming of T-cells by human monocyte-derived DCs, with inhibition of IFN-γ and enhancement of IL-4 production 65, that may underlie the association of allergic asthma with cigarette smoking. Conversely, endogenously released neutrophil elastase may inhibit maturation of murine DCs and inhibit the ability of mature DCs to present antigens to T-cells 66. Therefore, the function and role of DCs in COPD may be influenced by a multitude of external and host factors.
There is some evidence to suggest that CD1a+ mucosal-associated DCs may sustain CD8+ T-cell recruitment and retention in tissues 67, since CD1a+ DCs produce the ligands for CCR5 and CXC chemokine receptor (CXCR) 3, i.e. CCL3 and CXCL9, respectively. CD8+ T-cells also expressed a greater number of these receptors.
Monocytes/macrophages
Monocytes/macrophages are potentially very important effector cells in COPD through their potential release of ROS, extracellular matrix proteins and lipid mediators, such as leukotrienes, prostaglandins, cytokines, chemokines and MMPs 68. CD68+ macrophages are prominent and increased in number in the bronchial submucosa of COPD patients 39, 41, 69, and their numbers increase with increasing severity 8. Clusters of macrophages have been found particularly around small airways associated with the peribronchiolar fibrosis seen in smokers and ex-smokers 70. These may be associated with both the small airway fibrosis and centrilobular emphysema observed in COPD. Alveolar macrophages (AMs) in culture from smokers and COPD patients release increased amounts of MMP-1 and -9 71 with increased immunoreactivity 72, 73. Basal release of MMP-9 is increased, as is lipopolysaccharide-stimulated release of IL-6 and MCP-1 in COPD monocytes 74.
Microarray analysis of AMs from smokers with normal lung function and nonsmokers has shown increased expression of MMP-12, CCL2, CCR5 and osteopontin 75, 76, indicating the immune/inflammatory potential of these cells in smoking patients.
Macrophage proliferation occurs in smokers’ AMs since there is increased expression of the anti-apoptotic long isoform of B-cell leukaemia/lymphoma (Bcl)-X and increased cytoplasmic expression of the cyclin-dependent kinase inhibitor p21CIP/WAF−1, an inhibitory regulator of the cell cycle 77, and may, therefore, account for the increased number of macrophages in smoking and COPD. Increased expression of the proliferation marker, Ki67, in macrophages of smoking patients also supports the proliferative state of these cells 78. In addition, a defective phagocytic function towards apoptotic epithelial cells (termed efferocytosis) has been reported in COPD 79, 80. Kirkham et al. 81 showed that exposure of AMs to cigarette smoke impaired the ability of AMs to phagocytose apoptotic neutrophils, through the sequestration of receptors involved in the uptake of apoptotic cells and increased adhesion of macrophages to the matrix. Hodge et al. 78 showed that there is a reduction in the expression of several recognition molecules on AMs, namely CD44, CD71, CD31 and CD91, and a greater number of undifferentiated macrophages expressing lower levels of CD14 and with reduced phagocytic potential, which may explain the defect in efferocytosis in COPD. A previous report confirmed that a higher percentage of AMs from COPD patients express CD44, but with low surface expression 82. Defective clearance of apoptotic cells, such as neutrophils or epithelial cells, may result in secondary necrosis, which would exacerbate the inflammatory response in COPD 83.
EPITHELIAL CHANGES
The epithelial response to cigarette smoke may represent attempts by the airway epithelium to protect itself and repair the injury caused by cigarette smoke 84. Injury may lead to the development of squamous metaplasia, which is the reversible replacement of the columnar epithelium by squamous epithelium, an effect that has been correlated with airflow obstruction 85. Squamous cell metaplasia impairs mucociliary clearance and contributes to the increased risk of squamous cell carcinoma in COPD 86.
Several studies have examined gene expression in epithelial cells obtained by brushing from smokers and nonsmokers 87–90. The study of epithelial cells of small airways showed the upregulation of the pro-apoptotic gene pirin, antioxidant genes such as glutathione peroxidase, and ubiquitin carboxyl-terminal hydrolase (UCH) L1, a member of ubiquitin proteasome pathways, whereas the expression of IL-4 receptor, CX3C chemokine ligand (CX3CL) 1 and the extracellular matrix protein spondin 2 were inhibited 88. Conversely, genes upregulated following smoking that were downregulated with smoking cessation included UCHL1, trefoil factor 3, calcium-binding tyrosine-(Y)-phosphorylation-regulated protein, CXCL6, CX3CL1 and S100 calcium-binding protein A9 (S100A9); partial reversibility was shown by mucin (MUC) 5 subtypes A and C, and an irreversible gene was glycogen synthase kinase 3β 91.
Apart from regulation at the gene level, effects at the protein level have been reported. A rare proteomic study of lung samples from chronic cigarette smokers demonstrated upregulation of several proteins of the unfolded protein response (UPR), a mechanism compensatory to the interference of ROS with protein folding. These UPR proteins include chaperones, glucose-regulated protein 78, calreticulin and enzymes involved in antioxidant defences 92.
Small airway squamous metaplasia, as measured by expression of involucrin in stratified epithelium, increases with COPD severity 93; increased IL-1β expression by squamous cells has recently been postulated to lead to integrin-mediated activation of transforming growth factor (TGF)-β and, thus, amplification of pathological epithelial–mesenchymal interactions in COPD 12. Significant changes in oxidant responsive genes have been observed in the epithelium of healthy smokers as well as COPD patients 94. A small increase in the proliferative rate of the epithelium of small airways of COPD patients has been reported 95, together with increased expression of galectin-1 in the epithelium, which could be involved in epithelial proliferation and apoptosis.
Cigarette smoke induces the release of IL-1 and -8 and granulocyte colony-stimulating factor (G-CSF) from bronchial epithelial cells through oxidative pathways 96, accounting for potential neutrophil and monocytic chemotactic activities released from the epithelium 97. Higher expression of CCR2/MCP-1, TGF-β1 and CXCL8/IL-8 mRNA and protein has been observed in bronchiolar epithelium of smokers with COPD compared with smokers without COPD 98–100. Cultured epithelial cells from smokers and COPD patients release more TGF-β in vitro than do those from normal subjects 99. In addition, the expression of fibroblast growth factors 1 and 2 is increased in the bronchial epithelium of COPD patients 101.
Activation of the epidermal growth factor receptor (EGFR) cascade is increased in bronchial biopsy specimens from smokers with or without COPD compared with nonsmokers 102, 103, and smoking cessation does not lead to a reduction in EGFR expression 104. This indicates that chronic cigarette exposure may lead to permanent changes in the epithelium. Overexpression of EGFR has been one of the earliest abnormalities found in smokers at high risk of developing lung cancer 105, and somatic mutations acquired through smoking can persist for years 106, 107. The potential for these acquired somatic mutations in the molecular pathogenesis of COPD has been discussed elsewhere 108.
GOBLET CELLS, SUBMUCOSAL GLANDS AND MUCUS PRODUCTION
Goblet cell hyperplasia is more pronounced in smokers with COPD than in those without COPD 109. It contributes to mucus hypersecretion, which has been associated with morbidity and mortality 110, 111. Although an earlier study indicated no predictive value of mucus hypersecretion as regards mortality in COPD 112, others have associated chronic sputum production with the risk of hospitalisation, an excessive annual decline in FEV1 and the development of COPD 113. The increase in mucus production and reduction in mucociliary clearance caused by cigarette smoking represent an innate host defence response to this external attack. Post mortem study of lungs from patients who had died of COPD showed an increased amount of intraluminal mucus in the bronchioles compared with controls without respiratory disease 114. In surgically resected lung tissues, increasing accumulation of inflammatory exudates with mucus in the small airways has been noted with increasing severity of disease 8. Submucosal gland hypertrophy is also seen in the large airways 115, 116. A disproportionate increase in mucous acini and reduction in serous acini have been reported in chronic bronchitis 117, but there is no correlation between the mucous gland enlargement and sputum production 118, 119.
Goblet cell hyperplasia is a feature of both large and small airways in chronic bronchitis 115. Goblet cells are usually sparse in the small airways, but are present in increased number in small airways (diameter of <1 mm) of patients with COPD 120. Increased expression of MUC5B in the bronchiolar lumen and MUC5AC in the bronchiolar epithelium has been reported 121. In the large airways, there is increased stored MUC associated with an increase in MUC5AC expression accompanied by reduced MUC5B expression 109. Goblet cells have been associated with neutrophilic inflammation, supporting the concept that neutrophils, through the release of neutrophil elastase and cathepsin G, may directly cause degranulation of goblet cells 122. The mechanism of goblet cell hyperplasia itself may involve the activation of EGFR, which may be upregulated by oxidants in cigarette smoke and release of cytokines, such as tumour necrosis factor (TNF)-α and CXCL8/IL-8 and -13 123, 124. The downstream effects of IL-13 may require the activation of mitogen-activated protein kinase (MAPK) kinase/extracellular signal-regulated kinase (ERK), p38 MAPK and phosphatidylinositol 3-kinase (PI3K) 125, in association with the upregulation of a calcium-activated chloride channel that is specific to goblet cells in COPD 126, 127.
The regulation of MUC glycoproteins in airways diseases such as COPD has been reviewed. MUC genes are regulated by inflammation and respiratory pathogens 128. Cytokines and lipid components of Gram-positive or -negative bacteria activate MAPK pathways that may converge on Ras to activate nuclear factor (NF)-κB, specificity protein 1 or activator protein (AP)-1 transcription factor regulation of MUC2 or MUC5AC in the airways.
AIRWAY SMOOTH MUSCLE
A significant increase in airway smooth muscle in small airways of patients with COPD has been reported in several studies 129–131, and the amount of airway smooth muscle has been inversely correlated with lung function (FEV1 % predicted) 130. The amount of airway smooth muscle was increased by nearly 50% in patients with more severe COPD, at GOLD stages III and IV 8. In one study, the airway smooth muscle mass in the small airways was the only differentiating feature when comparing nonobstructed patients with COPD with patients with asthma 132. Although the airway smooth muscle mass is increased, it is not known whether this is caused by an increased number of airway smooth muscle cells, an increase in airway smooth muscle cell size or both. Smooth muscle protein isoform levels were not increased and, although the myosin light chain kinase level was slightly increased, there was no increase in phosphorylated myosin light chain levels 133. No increase in proliferation rate was observed in biopsy specimens obtained from patients with emphysema 134.
Airway smooth muscle cells not only have contractile properties but also are capable of expressing and releasing cytokines, chemokines, growth factors and proteases 135, 136, and can participate in the inflammatory and remodelling process 137. Airway smooth muscle cells also produce matrix proteins, and their behaviour may depend upon interactions with their own matrix 138. Cytokines and chemokines of potential interest in COPD, which may be released from airway smooth muscle, include IL-6 and CXCL8/IL-8, CCL2/MCP-1, -2/CCL8 and -3/CCL7, CXCL1/growth-related oncogene (GRO)-α, CXCL10/IFN-γ-inducible protein (IP)-10 and granulocyte-macrophage colony-stimulating factor (GM-CSF) 135, 139–141. The pro-inflammatory cytokines IL-1β, TNF-α and bradykinin induce the release of CXCL8/IL-8 140, 142, a potent neutrophil chemoattractant and activator, whereas IFN-γ and TNF-α induce the release of CXCL10/IP-10, which is also expressed in airway smooth muscle cells of patients with COPD 139. CXCL10/IP-10 is a potent chemoattractant for human monocytes, neutrophils, natural killer cells and T-cells, preferentially Th1 cells. Thymic stromal lymphopoietin is expressed in airway smooth muscle of patients with COPD 143 and may trigger DC-mediated Th2 inflammatory responses. Finally, airway smooth muscle cells are an important source of connective tissue growth factor (CTGF) and TGF-β 144, 145.
EXTRACELLULAR MATRIX CHANGES
Subepithelial basement membrane thickness is not usually increased in COPD as it is in asthma 34, 133, except in a subset of patients with reversible airways obstruction in whom an increase is observed in association with tissue eosinophilia. The increase in extracellular matrix is more diffuse throughout the airway mucosal surface, with an increase in total collagen I and III in the surface epithelial basement membrane, bronchial lamina propria and adventitia 146, together with an increase in laminin α2 in airway smooth muscle cells. There is also an increase in matrix deposition in the adventitial compartments of the small airways 8, 85, 147, and the fibrosis is characterised by an accumulation of fibroblasts and myofibroblasts. The presence of fibrillar collagen raises the possibility that the collagen is contracted and leads to fixed airflow limitation by preventing the complete relaxation of airway smooth muscle during hyperinflation or pharmacologically induced smooth muscle relaxation. Conversely, reduced expression of interstitial proteoglycans, such as decorin and biglycan, in the peribronchial area of small airways without any change in the expression of types I, II, and IV collagen, laminin or fibronectin 148 has also been reported. These conflicting data indicate the need for further studies on changes in the extracellular matrix in COPD and their relationship with airflow obstruction.
EMPHYSEMA AND FIBROSIS
Emphysema is characterised by the enlargement of alveolar spaces associated with destruction of alveolar walls, but without evidence of the fibrosis that may paradoxically occur in the small airways. Mechanisms for emphysema of COPD are listed in table 1⇓.
Mechanisms of emphysema in chronic obstructive pulmonary disease
Destruction of lung elastin has been put forward as a leading mechanism of alveolar destruction in the lungs, and release of neutrophil elastase and metalloproteinases from inflammatory cells, such as neutrophils and macrophages, may overwhelm the antiprotease defences of the lung and induce destruction. The accumulating evidence from animal models is reviewed below. In addition to this, there is indication that lung repair, as evidenced by de novo synthesis and tissue accumulation of elastin and collagen, is inhibited by cigarette smoke 149. Exposure to cigarette smoke extract also inhibits fibroblast proliferation 150, and fibroblasts isolated from patients with emphysema exhibit decreased proliferative capacity 151, 152. Exposure to cigarette smoke extract induces cell cycle arrest in fibroblasts, mediated through activation of p53 and p16, which inhibit the cell cycle, leading to cellular senescence 153, which may represent a response of fibroblasts to DNA damage by cigarette smoke extract 154. This may result in abnormal wound healing and prevention of repair of lung injury. In addition, cigarette smoke can kill endothelial cells and endothelial cell precursors 155, 156 and inhibit airway epithelial cell chemotaxis and proliferation 157. Cigarette smoke effects on alveolar epithelial cells also induce all the characteristics of senescence, including senescence-associated β-galactosidase activity, senescence-associated changes in cell morphology, an increase in cell size and lysosomal mass, accumulation of lipofuscin, overexpression of the cyclin-dependent kinase inhibitor p21CIP1/WAF1/Sdi1 and irreversible growth arrest 158. In mice exposed to cigarette smoke, activation of p21, through inhibition of p21-activated kinase, leads to oxidative and inflammatory responses and airspace enlargement 159. Loss of telomerase activity, a marker of senescence, has been reported in circulating lymphocytes of smokers with COPD 160. Table 1⇑ summarises the mechanisms that have been proposed for the development of emphysema.
An interesting contrast is that cigarette smoke, which inhibits fibroblast repair, contributing to lung tissue damage, is also associated with peribronchiolar fibrosis. This may be related to the production of growth factors released in the wall of the small airways. Some indication of the genes involved can be obtained from microarray studies of lung tissues from COPD patients. Genes involved in extracellular matrix synthesis and degradation and apoptosis were among the upregulated genes, including urokinase, urokinase receptor and thrombospondin in AMs and airway epithelial cells 161, involved in activation of TGF-β1 and metalloproteinases. Another microarray analysis of lung tissue from GOLD stage II patients found increased expression of the growth factors TGF-β1 and CTGF, with a reduction in the expression of collagen type I 162. Increased TGF-β, with possible activation of MMP-12, could lead to elastolytic effects with fibrotic effects, depending on its localisation in the airway or in the parenchyma. Cigarette smoke can result in activation of latent TGF-β, with stimulation of fibrosis 163. There are several mechanisms by which latent TGF-β can be activated in COPD; these include mediation by thrombospondin-1 164, integrins αvβ6 and αvβ8, which bind to arginine-glycine-aspartic acid sequences of TGF-β1 and TGF-β3 165, 166, serine proteases such as neutrophil elastase 167, and ROS.
APOPTOSIS OF LUNG CELLS
The role of apoptosis in COPD has been studied in lung tissue sections. An increase in endothelial cell apoptosis in lung tissue has been described, also with increased numbers of apoptotic alveolar epithelial cells, interstitial cells and inflammatory cells 72. Alveolar epithelial and endothelial cell apoptosis was also increased in other studies of emphysematous lung tissue 168–170, associated with an increase in activated subunits of caspase-3 and loss of the anti-apoptotic protein Bcl-2 169. Kasahara et al. 168 report that expression of vascular endothelial growth factor (VEGF) and VEGF receptor 2 protein and mRNA was significantly reduced in emphysema and, since these are maintenance factors for endothelial cells, a reduction may lead to endothelial alveolar septal death. Markers of oxidative stress and apoptosis, such as activated caspase-3, were co-localised in the central portion of the alveolar lobule in emphysema caused by VEGF receptor blockade. Inhibition of oxidative stress by a superoxide dismutase (SOD) mimetic inhibited alveolar cell apoptosis and emphysema in this model 171. VEGF levels in induced sputum from COPD patients decreased with the severity of the disease 172. In addition to apoptosis, increased cell proliferation has been reported in emphysema, presumably as a counterbalance to the increased apoptosis 169, 170.
CYTOKINES AND CHEMOKINES
Cytokines and chemokines are involved in many aspects of disease processes in COPD, including recruitment of neutrophils, macrophages, T-cells and B-cells, airway wall remodelling, including goblet cell metaplasia and epithelial cell hyperplasia, and the induction of emphysema.
Systemic cytokines/chemokines
Although systemic cytokines and chemokines may be considered to be produced locally, measurement of increased levels of cytokines in the circulating blood indicates a systemic component of the inflammatory process in COPD. This is in line with the clinical association of COPD with metabolic abnormalities, weight loss, muscle weakness and wasting, cardiovascular disease, depression, osteoporosis, cancer and anaemia 173. Evidence of systemic inflammatory processes in COPD can be judged from increased levels of cytokines, such as IL-6, CXCL8/IL-8, TNF-α and 55- and 75-kDa TNF receptor, together with C-reactive protein (CRP) levels 174. A recent study of severe-to-very-severe COPD showed that reduced FEV1 was associated with systemic inflammation, as measured by increased plasma levels of IL-6 and CRP; the elevated CRP was associated with decreased exercise endurance and poorer health status 175. Inflammation-associated oxidative stress, leading to skeletal muscle apoptosis, dysfunction and wasting, may account for the relationship between inflammation and loss of lean body mass in COPD 176. The source of these circulating cytokines may represent a spillover from pulmonary sources, but may also originate from other sources, such as circulating blood monocytes 177, striated muscle or concomitant atherosclerotic lesions. In stable COPD patients with respiratory muscle impairment, increased muscle expression of TNF-α and IL-6 gene and protein has been reported 178.
Lung cytokines/chemokines
The role of cytokines in COPD has been reviewed previously 179. Many reports describe elevated levels of CCL1/I-309, CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL11/eotaxin, CXCL1/GRO-α, CXCL5/epithelial neutrophil-activating peptide (ENA)-78, CXCL8/IL-8, IL-1β, IL-6, GM-CSF and TNF-α, measured in either induced sputum or BAL fluid or released from AMs exposed to cigarette smoke in patients with COPD 73, 180–186. Sputum neutrophil counts and level of CXCL8/IL-8 and circulating levels of TNF-α and CRP were the best markers relating to the severity of COPD 187.
The increased expression of CCL2 and its receptor CCR2 indicates that they may be involved in the recruitment of monocytes and immature DCs into the airways in COPD 98. However, another chemokine ligand, CCL20, the most potent known chemoattractant for DCs, is also upregulated in lung and induced sputum in COPD 59. This could account for the increased number of DCs observed in the small airways in COPD, since these cells also express CCR6, which is the receptor for CCL20.
Cigarette smoke increases CXCL8/IL-8 gene expression and release by bronchial epithelial cells, and that of TNF-α and IL-6 by AMs 96, 188, through oxidant mechanisms that include activation of the transcription factor NF-κB 189. Exposure of lung epithelial cells to smoke extract causes the release of neutrophil and monocytic chemotactic activities, with CXCL8/IL-8 and G-CSF accounting for the neutrophilic activity and CCL2/MCP-1 for the monocytic activity 97.
S100s or myeloid-related proteins are a family of calcium-binding proteins that are expressed in the cytosol of neutrophils, monocytes and macrophages. S100A9 concentration in BAL fluid is increased in COPD patients compared with asymptomatic smokers, as is S100A8 concentration in the BAL fluid of asymptomatic smokers versus nonsmokers 190. S100A8/14 have been shown to be endogenous factors in lung secretions that can stimulate IL-8 production by airway epithelial cells 191. S100A8/9 can directly induce neutrophil chemotaxis and adhesion 192.
Microarray analysis of lung tissues from COPD patients revealed the increased expression of CX3CL1/fractalkine, which is involved in monocyte emigration through tethering and arrest. Increased expression of CCL2/MCP-1, TGF-β1 and CXCL8/IL-8 mRNA and protein has been observed in bronchiolar epithelium, and of CCR2 in the macrophages of smokers with COPD compared with those of smokers without COPD 98. Since CCL2/MCP-1 binds to CCR2 and can induce T-cell and monocytic migration, CCL2 193 may contribute to the recruitment of these cells in COPD. Different chemokines were found to be involved in the chemotaxis of monocytes in induced sputum from COPD patients. Thus, there was increased chemotaxis of monocytes in COPD for CXCL1/GRO-α or CXCL7/neutrophil-activating peptide-2 but not for CXCL8/IL-8 or CXCL5/ENA-78, possibly due to differential regulation of CXCR2 receptors in COPD 194.
CD8+ and CD4+ T-cell-derived cytokines
The increased numbers of CD8+ T-cells expressing IFN-γ that infiltrate the peripheral airways of smokers with COPD show increased expression of the chemokine receptor CXCR3, which is paralleled by strong epithelial expression of its ligand, CXCL10/IP-10 48, a chemoattractant for Th1 cells. CCR5-positive T-cell numbers are increased in the airways of COPD patients with mild-to-moderate disease 195.
Although the above data would suggest that lung T-cells in COPD may be predominantly type-1-cytokine-producing CD8+ T-cells, there is evidence of expression of type-2 cytokines, but less prominently in COPD compared with asthma. Although IL-5 expression is absent in COPD airways associated with eosinophilia, other eosinophil chemoattractants, such as eotaxin or RANTES, may be implicated, particularly during exacerbations 196. Increased IL-4-positive cell numbers in mucus-secreting cells and associated plasma cells of the airway mucosa of chronic bronchitis patients has been reported 30. Although IL-18 expression appears to be increased in pulmonary macrophages of COPD patients, IL-13 gene expression appears to be decreased in emphysematous lung 197. IL-9 mRNA is overexpressed in CD3+ T-cells in patients with COPD 28. However, an increase in IL-4- and -13-positive cells in the bronchial submucosa in chronic bronchitis has been reported 198.
Much of the evidence for a role of IL-13 in COPD comes from work in mice. Overexpression of the Th2 cytokine IL-13 in lungs of adult mice induces emphysema, mucus goblet cell hyperplasia and airway inflammation, with macrophages, lymphocytes and eosinophils, and increased MMPs, which are many of the features associated with COPD 199. The induction of emphysema was related to the release of metallo- and cysteine proteases 199. Interestingly, the pro-inflammatory cytokine IL-18 can induce lung inflammation and emphysema through the production of IL-13 but not of IFN-γ 200, despite IFN-γ also being capable of inducing emphysema in this model 201.
EXACERBATIONS OF COPD: AMPLIFICATION OF LUNG INFLAMMATION
Exacerbations of COPD are particularly common in patients with advanced COPD, and are commonly associated with viral or bacterial infections 202, 203. Infections of the airways are important factors in enhancing the inflammatory response, providing an important trigger for further activation of inflammatory and immune cells already present and primed in the airways of patients with particularly advanced COPD. The defective ability of macrophages of COPD patients to phagocytose apoptotic neutrophils or epithelial cells also contributes, since a reduction in the clearance of infected cells leads to cell necrosis, with release of tissue-damaging products. These macrophages are also defective in phagocytosing bacteria 204. Clinical isolates of noncapsular Haemophilus influenzae may be ingested by neutrophils, which they can kill by necrosis 205. Viruses and bacteria may directly activate NF-κB and the MAPKs, leading to the release of inflammatory mediators and cytokines 206. Activation of NF-κB in lung macrophages obtained during an exacerbation has been reported 207.
Increases in plasma levels of IL-6, CXCL8/IL-8 and leukotriene B4 have been reported during COPD exacerbations requiring hospitalisation 208. During exacerbations of COPD associated with neutrophilia requiring ventilation, gene expression of CXCL5/ENA-78, CXCL8/IL-8 and the receptor CXCR2 is increased in tissue, particularly in the bronchiolar epithelium 209. Both CXCL5/ENA-78 and CXCL8/IL-8 bind to CXCR2 and are neutrophil chemoattractants. Eosinophils become more prominent among the cells recovered in sputum or bronchial biopsy specimens 33. In patients with an exacerbation of chronic bronchitis, CCL5/RANTES mRNA expression was upregulated and strong on the surface epithelium and subepithelial lymphomononuclear cells, together with increased numbers of eosinophils 196. CCL11/eotaxin and its receptor, CCR3, are upregulated during exacerbations of chronic bronchitis, with CCR3 mainly co-localised to eosinophils 210. Therefore, CCL11/eotaxin and CCL5/RANTES may underlie the eosinophilia of COPD exacerbations.
ANIMAL MODELS OF COPD FROM CIGARETTE SMOKE EXPOSURE
Development of animal models of COPD is essential for the testing of hypotheses derived from observation of the pathology of the disease and determining the mechanisms operating in COPD. Small rodents (guinea pigs, rats and mice) have also been exposed to cigarette smoke on a chronic basis, and there has been development of some features of COPD, namely inflammation and emphysema 211.
Variability of response in mice
The development of emphysema-like lesions is strain-dependent in the mouse 212, 213, and rats develop less emphysema than mice. Enlarged alveolar spaces and increased alveolar duct area are found after 3–6 months of tobacco smoke exposure in susceptible strains, such as B6C3F1 mice 214. The length of time required to produce emphysema is usually >4 months, depending upon the method of exposure, cigarette dose and species 215–217. At these later time points, tissue destruction seems to be mediated via macrophages. At the cellular level, neutrophil recruitment has been reported to occur immediately following the beginning of tobacco smoke exposure, and is followed by accumulation of macrophages. The early influx of neutrophils is paralleled by connective tissue breakdown. The early-stage alterations in neutrophil influx and increase in elastin and collagen degradation can be prevented by pre-treatment with a neutrophil antibody or α1-antitrypsin 218. Differences in response to chronic cigarette smoke exposure can be illustrated by the difference in response of two mouse strains, DBA and C57BL/6J, which are both sensitive to oxidant stress 213. DBA/2 mice develop more uniform parenchymal dilatation more rapidly, with areas of fibrosis with TGF-β expression, whereas C57BL/6J mice show extensive goblet cell hyperplasia, with expression of MUC5AC and IL-4 and -13 in the airways. Strains of mice resistant and susceptible to the development of emphysema due to chronic cigarette smoke exposure are useful in studying the genomic basis of susceptibility to emphysema 212.
Rats versus mice
Rats are also often used as models of COPD. However, they appear to be relatively resistant to the induction of emphysema-like lesions. Using morphometry and histopathology to assess and compare emphysema development in mice and rats, significant differences were demonstrated 214. In B6C3F1 mice, many of the morphometric parameters used to assess emphysema-like lesions differed significantly between exposed and nonexposed animals. In contrast, in exposed Fischer 344 rats, only certain parameters differed significantly from nonexposed values. The alveolar septa mean linear intercept was increased at 7 and 13 months in both exposed mice and rats, indicating an enlargement of parenchymal airspaces. However, the volume density of the alveolar airspace was significantly increased only in exposed mice. The volume density of the alveolar septa was decreased in mice at both time-points, indicating damage to the structural integrity of parenchyma. There was no alteration in Fischer 344 rats. Morphological evidence of tissue destruction in the mice included irregularly sized and shaped alveoli and multiple foci of septal discontinuity and isolated septal fragments.
Goblet cell metaplasia/hyperplasia has been described in a murine model of chronic cigarette smoke inhalation, but this response was also strain-dependent 213. A study in Sprague–Dawley rats exposed for 34 weeks showed features very much consistent with COPD, including small airways inflammation, goblet cell hyperplasia, airway remodelling and emphysema, and also changes in gene expression in the lungs that showed similarities to those found in human COPD 219. Upregulated inflammatory gene sets included CXCL1, CXCR1, CX3CL1, CCL3/MIP-1α, MMP-12 and osteopontin, whereas the stress response genes that were upregulated included SOD2, thioredoxin reductase and γ-glutamylcysteine synthetase.
In the rat model, there is also a degree of corticosteroid resistance that develops, as observed in patients with COPD. Thus, the inflammatory response observed after exposure of rats to cigarette smoke for 3 days is not inhibited by pre-treatment with corticosteroids 220, resulting from modifications in histone deacetylase (HDAC) 2 by oxidative stress imposed by cigarette smoke that render corticosteroids ineffective 221.
Other models have been used to mimic part of the process of COPD 211. Panacinar emphysema has been reproduced by treatment with chemicals, particulates and proteinases, such as pancreatic elastase, papain and neutrophil elastase. Repeated endotoxin administration induces neutrophilic inflammation with macrophage activation and airspace enlargement. Exposure to particulates or pollutant gases, such as ozone or nitrogen dioxide, creates models of oxidative stress to the lungs and airways.
MECHANISMS OF CIGARETTE SMOKE-INDUCED DAMAGE IN ANIMAL MODELS
Gene targeting and transgenic mice have been used to study the role of specific genes, particularly in the emphysema component, as listed in table 2⇓ 222. Emphysema is defined by morphometric enlargement of alveoli.
Potential factors involved in emphysema from gene-manipulated mice
Cytokines
Conditional overexpression of IL-1β via Clara cell secretory protein promoter led to pulmonary inflammation, increased airspace enlargement, enhanced MUC production, airway fibrosis and increased MMP-9 and -12 levels 223. Similarly, overexpression of IL-18, an IFN-γ-inducing factor and a member of the IL-1 cytokine superfamily, led to severe emphysematous changes, with increased IFN-γ and IL-13, and chronic inflammatory changes characteristic of COPD 200. Conversely, using a knockout model of IL-18 receptor α, cigarette-induced inflammation and emphysema were inhibited 226. Furthermore, in smokers and COPD patients, IL-18 expression and its downstream targets were shown to be increased in pulmonary macrophages, extending the relevance of these studies in mice into COPD.
The role of TNF-α in COPD was tested by studying the effect of chronic cigarette smoke exposure on TNF-α receptor knockout mice, in which there was partial inhibition of MMP-9 and -12 elevation, neutrophil and macrophage recruitment, and alveolar enlargement 227. However, the trial of an anti-TNF-α antibody in moderate-to-severe COPD showed no symptomatic or spirometric benefit 252, perhaps indicating that reversal of established COPD would be more difficult than previously perceived.
Overexpression of IL-13 and IFN-γ using inducible conditional transgenes led to emphysema in mice 199, 201. Overexpression of IL-13 resulted in inflammation and lung destruction that was MMP-9- and -12-dependent. There was goblet cell hyperplasia and subepithelial collagen deposition. A complex role for TGF-β in COPD has also been reported. Mice deficient in integrin αvβ6 fail to activate latent TGF-β in the lungs, and mice develop macrophage-rich inflammation with excessive MMP-12 that leads to spontaneous development of emphysema with time 253. MMP-12 has been shown to underlie cigarette-induced emphysema in mice 215.
Ageing and senescence
Mouse models have indicated the importance of the interactions of ageing with COPD. In aged lungs, there is increased fragmented alveolar elastin, which may activate neutrophil elastase, which could cause alveolar cell apoptosis. Mice with deletion of the anti-ageing Klotho protein develop early-onset emphysema, with MMP-9 activation 246. Senescence marker protein (SMP)-30, which is a liver protein that is downregulated in ageing rats, protects against alveolar enlargement and prevents cigarette smoke-induced alveolar septal damage and the increase in apoptotic lung cell numbers 244. Lack of SMP-30 in knockout mice appears to enhance the lung oxidative stress induced by chronic cigarette smoke exposure that may lead to the activation of MMPs or inactivate antiprotease activity. Therefore, cellular senescense, which is a process of replication fatigue, could contribute to emphysema 254, and cigarette smoke induces several of the processes involved in senescence in fibroblasts 153.
Innate immunity
A toll-like receptor (TLR) 4 mutation in C3H/HeJ mice is protective against cigarette smoke-induced pulmonary influx of neutrophils, DCs and lymphocytes upon subacute cigarette smoke exposure, but not on chronic exposure 247. This is likely to be mediating the effects of oxidative stress, since the lung inflammation induced by the oxidant ozone is blocked in TLR2-/- and TLR4-/- mice 255. TLR4-/- mice also developed spontaneous emphysema in middle age, associated with an oxidant/antioxidant imbalance due to increased expression of reduced nicotinamide adenine dinucleotide phosphate oxidase 3 and elastin degradation 248.
Oxidative stress
The roles of oxidative stress factors in cigarette smoke have been well studied in mouse models, and a clear picture of the mechanism is gradually appearing, with its importance in terms of both inflammation and emphysematous response to cigarette smoke exposure 13. Studies in mice have indicated the crucial role of the transcription factor nuclear factor erythoid 2-related factor (Nrf) 2, which binds to the antioxidant response elements of many antioxidant genes that lead to upregulation of many gene products. Levels of nuclear Nrf2 increase in epithelial cells exposed to cigarette smoke 92, and this may underlie the upregulation of many antioxidant genes in the airway epithelium, such as SOD2, glutathione peroxidase 2 and peroxiredoxin, in chronic cigarette smokers 87, 94, 256. Type 2 pneumocytes from Nrf2 knockout mice demonstrate impaired growth and increased sensitivity to oxidant-induced cell death 257. Finally, in vivo, Nrf2 knockout mice show increased susceptibility to emphysema and lung inflammation following cigarette smoke exposure accompanied by excessive oxidative stress and increased apoptosis 236.
The importance of antioxidants in protecting against cigarette smoke-induced effects was demonstrated in Cu/Zn SOD overexpressing mice. No neutrophilic inflammation and a reduced onset of airspace enlargement was noted after a 1 yr exposure to cigarette smoke 242.
OXIDATIVE STRESS, TRANSCRIPTION FACTORS AND SIGNALLING PATHWAYS
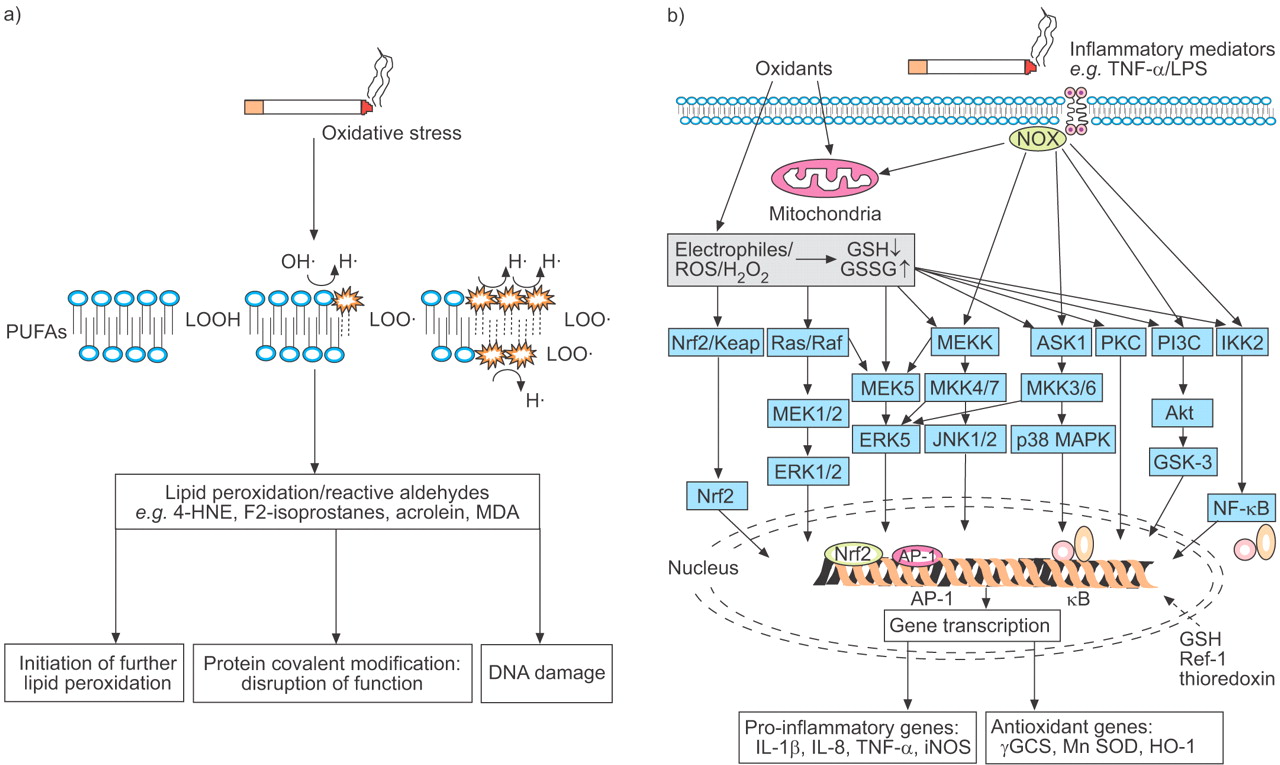
ROS can activate a number of redox-sensitive transcription factors, including NF-κB and AP-1, which may potentiate the inflammatory response in the lungs of COPD patients (fig. 3⇓). In addition, other signal transduction pathways, such as MAPKs and PI3K, are also activated by ROS stress 258–260. Using ozone exposure of small rodents, confirmation of activation of NF-κB, c-Jun N-terminal kinase (JNK) and p38 MAPK in lung cells was obtained 261, 262, and the importance of the MAPK signalling pathway in ozone-induced neutrophilic inflammation ascertained. Increased expression of the phosphorylated p38 MAPK in the airway cells of COPD patients, particularly in AMs and alveolar wall, has been reported 263. In tracheobronchial epithelium grown from primary human lung epithelial cells at an air–liquid interface, exposure to cigarette smoke led to the activation of MAPK pathways, including c-Jun, Jun D, ERK and p38, plus the expression of the EGFR ligand, amphiregulin and heparin-binding epidermal growth factor 264. A p38 MAPK inhibitor, SD-282, reduced short-term cigarette smoke-induced inflammatory increases in macrophage and neutrophil numbers in BAL fluid, an effect not mimicked by corticosteroids 265.

{kind=link}
{kind=link}
{kind=link}
Effects of oxidative stress arising from exposure to cigarette smoke on lung cells. a) Oxidative effects on membrane fatty acids leading to lipid peroxidation and reactive aldehydes. b) Intracellular activation of kinases and transcription factors that lead to gene transcription of antioxidants and pro-inflammatory genes. PUFA: polyunsaturated fatty acid; LOOH: lipid hydroperoxide; 4-HNE: 4-hydroxy-2-nonenal; MDA: malondialdehyde; TNF: tumour necrosis factor; LPS: lipopolysaccharide; ROS: reactive oxygen species; GSH: reduced glutathione; GSSG: oxidised glutathione; Nrf: nuclear factor erythoid 2-related factor; KEAP: Kelch-like erythroid-cell-derived protein with CNC homology-associated protein; MEK: mitogen-activated protein (MAP) kinase kinase; ERK: extracellular signal-regulated kinase; MEKK: MAP/ERK kinase kinase; MKK: MAP kinase kinase; JNK: c-Jun N-terminal kinase; ASK: apoptosis signal-regulating kinase; MAPK: MAP kinase; PKC: protein kinase C; PI3K: phosphatidylinositol 3-kinase; Akt: Akt kinase; GSK: glycogen synthase kinase; IKK: inhibitor of nuclear factor (NF)-κB kinase; AP: activator protein; Ref: redox factor; IL: interleukin; iNOS: inducible nitric oxide synthase; γGCS: γ-glutamylcysteine synthetase; SOD: superoxide dismutase; HO: haem oxygenase.
Oxidative stress and the redox status of the cells can also directly or indirectly regulate histone modifications, such as acetylation, methylation and phosphorylation, leading to enhanced induction of inflammatory mediators 13, 259, 260. In AMs and bronchial epithelial cells, ROS amplify the induction of inflammatory mediators, such as GM-CSF, CXCL8/IL-8, IL-1β, cyclooxygenase 2, nitric oxide synthase-2 and TNF-α, and antioxidant enzymes (glutamate cysteine ligase, Mn SOD and thioredoxin) 13, 266. This may account for the increased expression of inflammatory mediators reported in the BAL fluid of smokers 267, 268 and in epithelial cells in response to cigarette smoke treatment 269, 270. Furthermore, AMs from the lungs of smokers are more activated than those obtained from nonsmokers.
There is clear evidence for NF-κB activation in bronchial epithelial cells of patients with COPD and in sputum macrophages during exacerbations of COPD 207, 271. Nuclear localisation of p65 is enhanced within the bronchial epithelium of COPD patients and, to a lesser extent, in control smokers, in comparison with control nonsmokers 271. In lung tissue from patients with COPD, an increase in NF-κB nuclear translocation associated with degradation of the inhibitor of NF-κB (IκB)α is observed, as well as an imbalance between histone deacetylation and acetylation in favour of acetylation 272.
The mechanism by which ROS enhance NF-κB activation, however, may be cell specific and distinct from physiological activators, such as TNF-α and IL-1β, since diamide, which oxidises reduced glutathione to oxidised glutathione, and H2O2 are unable to activate NF-κB in certain cell types 273. H2O2 has been proposed to phosphorylate and directly activate IκB kinase 2 in HeLa cells 274, 275, and ROS stress can cause rapid ubiquitination and phosphorylation of the IκBα complex with subsequent degradation in some cell types 276, 277. Alternatively, oxidative stress may affect the proteasome enzymatic activity that leads to activation of NF-κB 275, 278. Finally, ROS may interfere with p65 activity independently of nuclear translocation by affecting the recruitment or activity of transcriptional coactivators, leading to increased local histone acetylation and enhanced gene transcription 13. An important characteristic of the inflammation in COPD is the reduced or lack of anti-inflammatory effects of glucocorticoids, which has been linked to oxidative stress 19, 279.
ROS may impinge upon several steps in the glucocorticoid receptor activation pathway. In COS-7 and Chinese hamster ovary cells, oxidative stress with H2O2 reduced nuclear transport 280. Nitrosyl stress induced by the nitric oxide donor S-nitroso-dl-penicillamine prevents glucocorticoid receptor dissociation from the heat shock protein 90 complex and a reduction in ligand binding 281. Lung tissues from patients with increasing severity of COPD showed graded reductions in HDAC activity, essential for the suppression of many inflammatory genes by glucocorticoids 282, and increases in IL-8 expression and histone-4 acetylation at the IL-8 promoter. HDAC2 expression and activity were decreased in patients with COPD with no changes in histone acetyltransferase activity 283. This results in reduced glucocorticoid responsiveness and increased histone acetylation, which, in turn, leads to an increase in inflammatory cytokine expression. This observation suggests a possible mechanism involved in the decreased efficacy of glucocorticoids in cigarette smokers.
This effect can also be mimicked in vitro and ex vivo in epithelial cells and macrophages with H2O2 or the nitric oxide donor 3-morpholinosydnonimine, and parallels the effects seen with the HDAC inhibitor trichostatin A 284. Furthermore, the antioxidant N-acetyl-l-cysteine was able to prevent the enhanced cytokine release and partially restore dexamethasone sensitivity to these cells 285. This suggests that oxidative stress caused by repression of HDAC activity can modulate glucocorticoid receptor function in macrophages. In a rat model of cigarette smoke-induced lung inflammation, Marwick et al. 220 showed an influx of inflammatory cells and induction of histone modifications in the lungs. This was associated with increased NF-κB, AP-1 and p38 MAPK activation and increased histone 4 acetylation. Decreased HDAC2 activity, related to protein modification by aldehydes and nitric oxide products, was also observed and correlated with a lack of corticosteroid suppression of smoke-induced pro-inflammatory mediator release 220. Redox-sensitive post-translational modifications of HDAC1–3 are associated with loss of activity and enhanced inflammatory mediator release in a human macrophage cell line 286. Nicotinamide adenine dinucleotide-dependent deacetylase sirtuin (SIRT)-1, which belongs to the class III HDACs, interacts with the p65 subunit of NF-κB to regulate cigarette smoke-mediated pro-inflammatory cytokine release, implicating a role for SIRT1 in sustained inflammation and ageing of the lungs 287. SIRT1 levels are reduced in macrophages and lungs of smokers and patients with COPD, an effect that has been related to post-translational modifications by cigarette smoke-derived oxidants 288.
Although not examined in COPD, changes in the activation status of various kinase pathways have been associated with reduced glucocorticoid responsiveness in severe asthma 289, 290. Thus, enhanced activation of ERK, JNK and p38 MAPKs and signal transducers and activators of transcription has been proposed to play a role in steroid-insensitive asthma 291–295.
CONCLUSIONS
Since the beginning of the 2000s, an increase (if not an explosion) has been seen in the number of publications on the pathophysiology and mechanisms underlying chronic obstructive pulmonary disease. In addition to understanding the processes of inflammation and tissue destruction and degradation, other cellular and molecular processes, such as autoimmunity, apoptosis of alveolar cells and senescence, have become implicated in chronic obstructive pulmonary disease. These complicate the picture, and new paradigms and mechanisms will emerge from the continuing study of tissues from chronic obstructive pulmonary disease patients and the use of animal models of cigarette exposure. The impact of coexisting diseases, such as cardiovascular diseases and cancer, can also contribute to the pathophysiology. Susceptibility to the development of chronic obstructive pulmonary disease is an unsolved mystery, and the contribution of these different mechanisms to the heterogeneity and outcome of chronic obstructive pulmonary disease needs to be evaluated. New specific treatments could be envisaged for specific mechanisms operating in particular chronic obstructive pulmonary disease patients and for specific subcategories of chronic obstructive pulmonary disease patient. Therefore, it is imperative to understand whether specific mechanisms lead to particular clinical phenotypes in this disease.
Statement of interest
None declared.
Footnotes
-
Previous articles in this series: No. 1: Oeckler RA., Hubmayr RD. Ventilator-associated lung injury: a search for better therapeutic targets. Eur Respir J 2007; 30: 1216–1226.
- Received February 7, 2008.
- Accepted February 15, 2008.
- © ERS Journals Ltd
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











Jump To
- Article
- Abstract
- STUDYING MECHANISMS OF COPD
- AIRWAY INFLAMMATION IN COPD
- EPITHELIAL CHANGES
- GOBLET CELLS, SUBMUCOSAL GLANDS AND MUCUS PRODUCTION
- AIRWAY SMOOTH MUSCLE
- EXTRACELLULAR MATRIX CHANGES
- EMPHYSEMA AND FIBROSIS
- APOPTOSIS OF LUNG CELLS
- CYTOKINES AND CHEMOKINES
- EXACERBATIONS OF COPD: AMPLIFICATION OF LUNG INFLAMMATION
- ANIMAL MODELS OF COPD FROM CIGARETTE SMOKE EXPOSURE
- MECHANISMS OF CIGARETTE SMOKE-INDUCED DAMAGE IN ANIMAL MODELS
- OXIDATIVE STRESS, TRANSCRIPTION FACTORS AND SIGNALLING PATHWAYS
- CONCLUSIONS
- Statement of interest
- Footnotes
- References
- Figures & Data
- Info & Metrics