





Abstract
The biology of O2 is extremely complex and defies a comprehensive yet brief summary that showcases its roles across the entire animal and plant kingdom. This necessarily short introduction to the 2006 Taormina Lung Science Conference (Taormina, Italy) on hypoxia will examine key features of the biology of O2 only within mammalian cells, even that being a daunting task.
SERIES “HYPOXIA: ERS LUNG SCIENCE CONFERENCE”
Edited by N. Weissmann
Number 1 in this Series
Traditionally, physiologists consider how O2 travels from the environment to the mitochondria, i.e. the transport processes and the limiting factors. Metabolists consider the role of O2 in energy generation via oxidative phosphorylation, i.e. both as substrate and regulator of cell processes, including its own utilisation. Biochemists consider, among other things, O2 as a source of reactive O2 species (ROS) and how both O2 (or lack thereof) and ROS may affect transcription of many genes. Pathologists and clinicians consider effects of both insufficient and excess O2 on tissue structure and function. Oxygen is at the centre of a remarkable, integrated system that scientists are just beginning to piece together, and the goal of the present introduction to the series is to describe how this multifaceted integration may occur.
O2 TRANSPORT
The lungs
For many years, respirologists’ interests in O2 focused mostly on understanding how O2 does (or does not) get from the air to pulmonary capillary blood. Stimulated originally by wartime needs ∼60 yrs ago, several generations of researchers 1–6 have developed the current physiological understanding of O2 transport to quite sophisticated levels with the coming together of new ideas and new techniques. A great deal is currently known about ventilation, blood flow, the distribution of one to the other, and gas exchange 6, 7. Scientists have been able to define when diffusion limitation of gas exchange occurs in the lungs, in both health and disease 8, 9, and have recognised that pulmonary gas exchange affects gas exchange in the tissues and vice versa, that is, the system is clearly integrated throughout the body.
The tissues
Compared with exchange at the lungs, less attention has been given to understanding how O2 delivered via the arterial blood supply of a tissue or organ does (or does not) get to the mitochondria in that tissue. Interestingly, essentially the same physiological barriers as in the lung potentially impede O2 transport within tissues: heterogeneity of physiological O2 supply versus biochemical demand for O2; diffusional transport limitation between tissue vascular red blood cells and mitochondria; reduced delivery via low blood flow, haemoglobin or O2 saturation; and shunting of blood around tissue elements that need it. These have proven much more elusive to characterise and thus understand than their pulmonary counterparts but, slowly, “in-roads” are being made into these processes as well. There is still a long way to go, especially in severe diseases marked by multiorgan failure. One system that has been evaluated is skeletal muscle. It is a useful model to demonstrate the integration that revolves around cellular oxygen tension (PO2).
Based on magnetic resonance spectrometry measurements of cytoplasmic myoglobin saturation 10, myoglobin is almost fully saturated at rest, implying that cytoplasmic PO2 must be fairly close to that of venous blood. However, intracellular PO2 falls during exercise to remarkably low but stable levels of ∼0.4–0.5 kPa (3–4 mmHg), indicated by a myoglobin O2 saturation of ∼50%. This low PO2, which represents the balance between O2 supply and O2 demand, has many remarkable implications and consequences and raises several questions as follows.
1. Should one look at such a low PO2 as hypoxia (a failure of supply to keep up with demand) or is this a regulated value (i.e. normoxia) that serves major biological purposes? Keeping in mind that mitochondria can operate at full speed at such low PO2 levels 11, it is easy to argue that a PO2 of 0.4–0.5 kPa (3–4 mmHg) is all that is needed to run the system. Indeed, more would be wasteful and might possibly predispose to excess generation of ROS and therefore, perhaps, to cellular O2 toxicity. In other words, the system may be regulated to a cell PO2 of 0.4–0.5 kPa (3–4 mmHg), which is advantageous.
2. Additionally, this low PO2 means that the PO2 gradient from red cells to mitochondria is maximised, favouring diffusive transport of O2, the only way for O2 to reach mitochondria. However, a system that needs the gradient maximised suggests that O2 transport from red cells to mitochondria is diffusion-limited, and there is much evidence to support this notion at maximal oxygen uptake 12–14. In turn, this implies that maximal O2 utilisation is limited by its own transport rather than by the capacity of the cells to use O2, for which there is also evidence 14–16.
3. The low PO2 is also important in local blood flow regulation, since local hypoxia dilates local tissue microvessels 17–19, resulting in the augmentation of flow when and where metabolic rate increases to help match O2 supply to O2 need. Therefore, it is easy to hypothesise that this low PO2 is regulated to optimise O2 transport.
4. As mentioned hereafter in more detail, low PO2 values lead to gene activation for systems that are upregulated in response to reduced PO2 in a manner designed to augment O2 availability. This happens through the hypoxia-inducible factor (HIF) system 20 but may also occur through other pathways. For example, a low PO2 enhances muscle expression of vascular endothelial growth factor (VEGF) 21, likely to occur (in part at least) via HIF-1α 22. Over time, this may lead to angiogenesis, which permits greater blood flow to support a greater metabolic rate. It is easy to speculate for muscle that, without a low intracellular PO2 during exercise, adaptive gene expression (i.e. exercise training) might not occur.
In summary, PO2 inside the myocyte falls dramatically during exercise due to multiple, quite different but complementary reasons: 1) to maximise diffusive O2 transport; 2) to regulate ROS generation; 3) to regulate local perfusion to serve local metabolic need; 4) to regulate the speed of metabolic reactions to match O2 availability; and 5) to stimulate adaptive gene expression to enhance future exercise.
O2 AND THE TISSUES: NOT GOOD TO HAVE TOO MUCH OR TOO LITTLE
In recent years, the study of O2 has taken some dramatic new turns. Rather than simply worrying about how O2 transport occurs, researchers have turned their attention to how O2 (and especially its reduction or excess stemming from altered transport) may affect cell, tissue and organ function; O2 is far more than a substrate for energy production (fig. 1⇓).

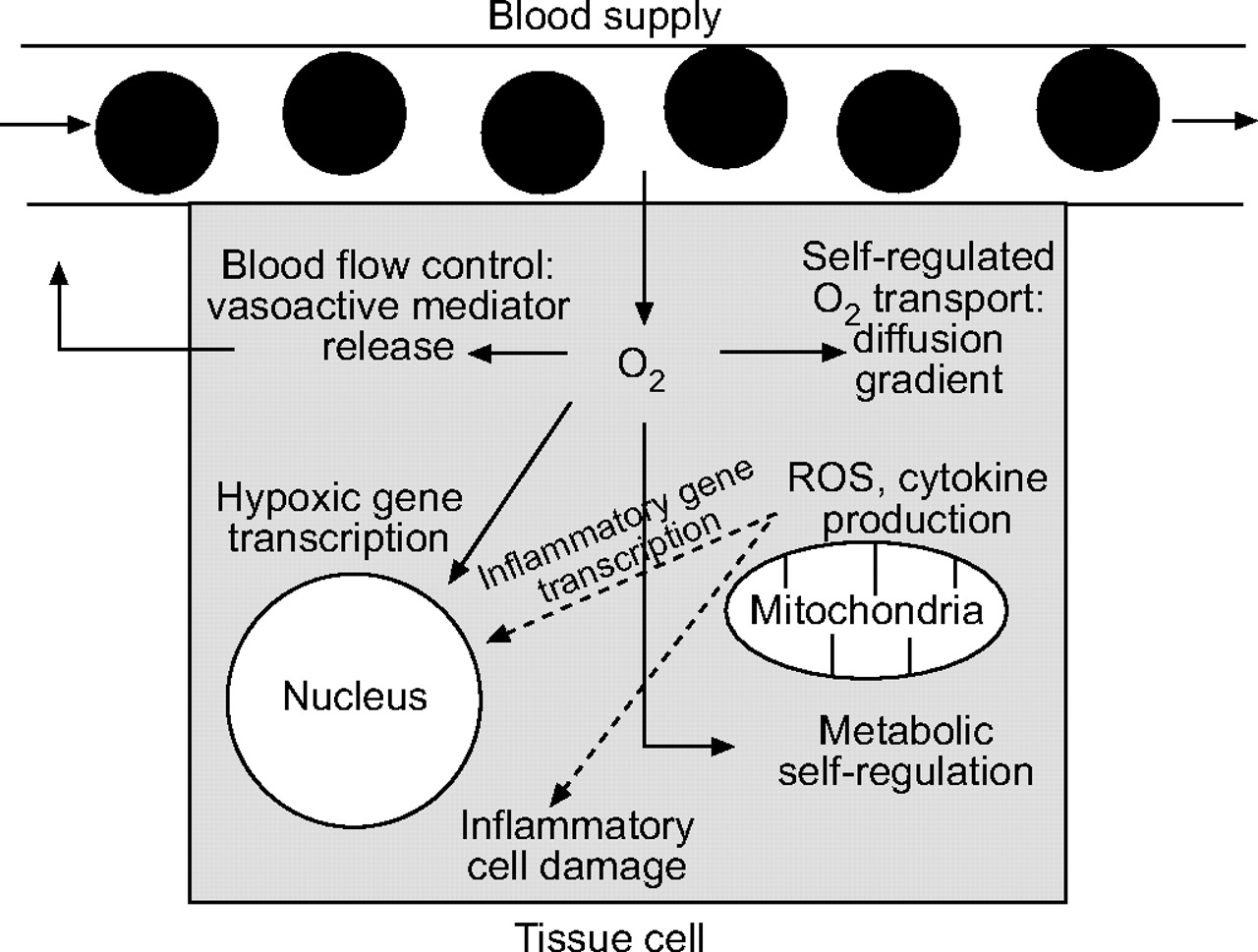{kind=link}
Multiple functions of O2 within tissue cells. While this scheme is very unlikely to describe all ways in which O2 influences cell structure and function, it serves to illustrate the multiple levels at which oxygen tension plays a role in cellular events such as transport, metabolism, inflammation and gene transcription. ROS: reactive oxygen species.
Researchers have known about excess O2 causing pulmonary O2 toxicity for many years. From initial physiological descriptions it is clear that excess O2 leads to high levels of ROS, inflammation and tissue damage. It has also been well known for many years that reduced O2 causes serious disturbances, such as tissue death in heart, brain, extremities, gut, etc. Hypoxic pulmonary vasoconstriction affects pulmonary circulation, ultimately contributing to potentially fatal illnesses, such as high-altitude pulmonary oedema 23. Researchers have recognised that acute hypoxia in peripheral tissues causes vasodilatation in a compensatory effort to restore O2 transport and that persistently reduced O2 (e.g. at altitude) during development and early post-natal life stimulates a number of characteristic changes, including increased lung volumes and blood O2 transport ability 24–26. Additionally, post-natally reduced O2 stimulates typical responses, the most evident of which are hyperventilation via chemoreceptor stimulation and erythrocytosis via erythropoietin, both apparently targeted to restore O2 availability.
Despite these restorative responses, the hypoxia of altitude produces progressively more extreme and deleterious effects on the body. While some individuals can make the 8,848 m ascent of Mount Everest without breathing supplemental O2 27, in spite of the fact that inspired PO2 on the summit is only 5.3–6.0 kPa (40–45 mmHg) 28 and arterial PO2 is only 3.3–4.0 kPa (25–30 mm Hg) 29, man cannot live and reproduce at altitudes ∼>5,000 m for more than short periods of time 30. Basically, every organ is affected by such degrees of hypoxia, with loss of lean body mass, and cerebral, gastrointestinal and reproductive disturbances that progress to intolerance for that altitude.
Even at sea level and in the absence of parenchymal disease per se, sleep-disordered breathing produces levels of hypoxia that lead to many responses considered to be disadvantageous to the organism. Cycles of hypoxia and re-oxygenation are reported to cause systemic inflammation, likely via ROS 31, clinically important dysregulation of the autonomic nervous system resulting in systemic hypertension 32, 33 and, it seems, predisposition to the metabolic syndrome 34.
O2-RELATED PATHOPHYSIOLOGY: MECHANISMS
As these phenotypic responses have been progressively catalogued over the years, research has begun to focus on how changes in O2 lead to changes in function, that is, the mechanisms. It is known that in many tissues, both high and low levels of O2 promote greater generation of ROS that may themselves lead to inflammation, signalling and gene activation. It is known that altered levels of O2 in many tissues affect gene expression directly. The overarching role of HIF in regulation of many genes associated with restorative responses to hypoxia has emerged as one of the major stories of recent years 20. The stabilisation of HIF by hypoxia 35, which provides enough of this factor to cause transcriptional activation of a whole series of genes whose proteins act to counterbalance the reduction in available O2, is now quite well understood. Prominent among HIF-regulated genes are those encoding glycolytic pathway enzymes, glucose transporters, erythropoietin and VEGF to name just four categories. Undoubtedly, the mechanisms will prove to be complex, far more so than this trivial description.
O2: HOW IT IS SENSED
How levels of O2 may be sensed to initiate the many consequences aforementioned remains a “hot topic” with evidence for various sensors 4, 36 at the mitochondrial level, within the HIF system, during the generation of ROS and possibly others. Interestingly, as the many O2-dependent pathways are better understood, it seems that the associated responses may not be fully explained by hypoxia alone. For example, it may be that O2-independent signalling pathways in muscle might be involved in activation of VEGF in concert with hypoxia.
O2 AND THE GENOME: CAUSE AND EFFECT
Understanding the details of these many, varied and complex signalling pathways remains a major challenge. But even when these are elucidated, there will still exist the need to determine their importance. Some have been established beyond doubt, but appropriate gene manipulation strategies will need developing to be certain about the importance of others. As an example, it is possible to discuss skeletal muscle. It has been found that VEGF is indeed the key angiogenic growth factor in muscle. In vivo deletion of this gene in the adult mouse by conditional knockout using Cre-LoxP techniques has demonstrated that two thirds of the muscle capillaries disappear in regions where VEGF is substantially absent 37. Over ≥8 weeks there is no recovery of capillarity and considerable apoptosis occurs. Similar results are seen with lifelong whole muscle VEGF deletion using a cross-breeding strategy to delete VEGF throughout from embryonic stages, but only in muscle 38. Remarkably, these findings suggest that no adequate parallel or compensatory pathway exists for this gene and tissue. Deletion of VEGF in the lung by similar methods also produces major effects 39, especially apoptosis and an emphysema-like phenotype, but whether this plays into clinical emphysema in humans remains to be determined.
SUMMARY
The objective of the present far-ranging introduction to the 2006 Taormina Lung Science Conference was to briefly cover some of the remarkable developments in the study of the biology of O2 over the past century: from physiological interests in its transport to how it acts as a signalling molecule; how it affects tissue structure and function when in high or low concentrations; and how such changes in availability affect genomic responses presumably designed to restore function under such circumstances. These many faces of O2 coexist of course as a remarkable, integrated system of transport, utilisation, signalling and pathogenesis.
This series will continue the discussion of the many faces of O2 and will lead the participants down new roads in the unending quest to understand its very complex biology.
Statement of interest
None declared.
- Received November 19, 2007.
- Accepted December 23, 2007.
- © ERS Journals Ltd
References










