





Abstract
Interleukin (IL)-18 production and pulmonary function were evaluated in patients with chronic obstructive pulmonary disease (COPD) in order to determine the role of IL-18 in COPD.
Immunohistochemical techniques were used to examine IL-18 production in the lungs of patients with very severe COPD (Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage IV, n = 16), smokers (n = 27) and nonsmokers (n = 23). Serum cytokine levels and pulmonary function were analysed in patients with GOLD stage I–IV COPD (n = 62), smokers (n = 34) and nonsmokers (n = 47).
Persistent and severe small airway inflammation was observed in the lungs of ex-smokers with very severe COPD. IL-18 proteins were strongly expressed in alveolar macrophages, CD8+ T-cells, and both the bronchiolar and alveolar epithelia in the lungs of COPD patients. Serum levels of IL-18 in COPD patients and smokers were significantly higher than those in nonsmokers. Moreover, serum levels of IL-18 in patients with GOLD stage III and IV COPD were significantly higher than in smokers and nonsmokers. There was a significant negative correlation between serum IL-18 level and the predicted forced expiratory volume in one second in patients with COPD. In contrast, serum levels of IL-4, IL-13 and interferon-γ were not significantly increased in any of the three groups.
In conclusion, overproduction of interleukin-18 in the lungs may be involved in the pathogenesis of chronic obstructive pulmonary disease.
- Chronic obstructive pulmonary disease
- chronic obstructive pulmonary disease clinical/basic investigations
- cytokine production
Chronic obstructive pulmonary disease (COPD) is an important pulmonary inflammatory disease of which the prevalence and associated mortality rates have been predicted to rise. Smoking is recognised as the largest risk factor for COPD, and quitting smoking is thought to be important in the prevention and control of COPD 1, 2. However, there is, as yet, no effective treatment for pulmonary inflammation in COPD.
Increased numbers of CD8+ T-cells, alveolar macrophages and neutrophils are characteristic pathological features of the lungs in COPD 3, 4. Eosinophil numbers are increased during exacerbations and possibly also during stable phases in a subset of patients 5. These activated inflammatory cells can release various mediators, including leukotriene B4 and inflammatory cytokines (e.g. T-helper cell (Th) types 1 and 2 cytokines, and tumour necrosis factor (TNF)-α) 6. The Th1 cytokine interferon (IFN)-γ may contribute to the onset of COPD 7. For example, previous studies have demonstrated positive correlations between CD8+ cell numbers and the degree of COPD airflow limitation 8. The CD8+ lymphocytes are believed to be predominantly cytotoxic T-cells type 1 that produce IFN-γ 7. One study 9 reported that the level of IFN-γ-inducible protein-10 (CXC chemokine ligand 10) was also increased in the lungs of COPD patients, but recent studies 10, 11 have reported that IFN-γ production in the lungs of these patients is not significantly greater than in healthy controls. In mice models, pulmonary emphysema has been induced by doxycycline in lung-specific IFN-γ transgenic (Tg) mice 12. The Th2 cytokines interleukin(IL)-4 and -13 are also thought to be involved in the pathogenesis of COPD. For instance, levels of IL-4 and -13 are significantly greater in the central airways of smokers with chronic bronchitis than in those of asymptomatic smokers 10. In contrast, a previous study 11 reported decreased expression of IL-13 in the lungs of COPD patients. Overproduction of IL-13 induced in the lungs of adult mice by a doxycycline-dependent Clara cell secretory protein promoter induces emphysema 13.
IL-18 is a proinflammatory cytokine produced intracellularly from a biologically inactivated precursor, pro-IL-18. Mature IL-18 is secreted after cleavage of pro-IL-18 by caspase-1, originally identified as IL-1β converting enzyme. Activated macrophages produce large amounts of mature IL-18 after cleavage of pro-IL-18 by caspase-1 14. It has been reported that low levels of pro- and mature IL-18 proteins are expressed in the lungs of healthy subjects 15. Hoshino and co-workers 16–18 have shown that IL-18 can act as a co-factor for both Th1 and Th2 cell development, and several studies have subsequently reported that IL-18 may be involved in the development of Th2-type-diseases, such as asthma 14. The present authors have previously demonstrated that treatment with recombinant IL-18 plus IL-2 induces pulmonary inflammation and lung injury in mice 19, and IL-18 and its receptor are strongly expressed in the human lung in idiopathic pulmonary fibrosis 15. Very recently, the present authors reported that constitutive IL-18 overproduction in the lungs of mice induces emphysema 20. Taking these results together, it is anticipated that IL-18 is involved in the pathogenesis of pulmonary inflammatory diseases, such as lung fibrosis, lung injury and COPD.
The aim of the present study was to analyse the expression of IL-18 in the lungs and sera of COPD patients in order to evaluate the role of IL-18 production in the pulmonary function of patients with COPD. Some of the results of the present study have been previously reported in the form of an abstract 21.
MATERIALS AND METHODS
Subjects
In total, 78 COPD patients (67 males and 11 females) were monitored at Kurume University Hospital (Kurume, Japan), Fukuoka University Hospital (Fukuoka, Japan), Chikugogawa Onsen Hospital (Ukiha, Japan), Kirigaoka Tsuda Hospital (Kitakyushu, Japan), Shigemoto Hospital (Shimonoseki, Japan), Keisinkai Hospital (Tosu, Japan), Tokunaga-Naika Clinic (Fukuoka, Japan), the Social Insurance Futase Hospital (Iizuka, Japan) and Arao Central Hospital (Arao, Japan). All patients with COPD were diagnosed on the basis of clinical history, physical examination, chest radiograph, chest computed tomography and pulmonary function tests in accordance with the Global Initiative for Chronic Obstructive Lung Disease (GOLD) clinical criteria for the diagnosis and severity of COPD 22. Exclusion criteria were: chronic lung conditions, such as asthma, bronchiectasis and interstitial lung diseases; cardiac, hepatic and renal failure; and current oral steroid therapy. Lung tissues were obtained from 16 patients with very severe COPD (GOLD stage IV) who had undergone lung volume reduction surgery (LVRS) at Fukuoka University Hospital. Control lung tissues were obtained from the normal tissues around preserved cancer specimens obtained after surgery from 23 nonsmokers and 27 smokers who underwent lung cancer extirpation at Kurume University Hospital. No patient was able to be sampled for both lung tissues and serum. Lung diseases (e.g. sarcoidosis, infectious diseases) were carefully excluded in control subjects, and ex-smokers were carefully excluded from the group of nonsmokers. Informed written consent was obtained from each subject and sample collection and all procedures were approved by the ethics committees of Kurume University and Fukuoka University.
Pulmonary function tests
Predicted normal Japanese values were used to calculate the forced expiratory volume in one second, (FEV1) % predicted, which met the Japanese Pulmonary Function Standard in the Japanese Respiratory Society Statement 23. Further details of these analyses are provided in the online supplementary data.
Histology
Samples of lung tissue were fixed with 10% formalin and embedded in paraffin wax. Between one and three paraffin-embedded lung tissue samples were obtained from each subject. Sequential sections were made from each paraffin-embedded lung tissue. Sections (4 μm thick) were cut and placed on poly-l-lysine coated slides and then incubated overnight at 55–60°C. Deparaffinised sections were stained with haematoxylin and eosin (HE).
Morphometric analysis
The cross-sectional area occupied by the wall was quantified as a ratio of the total cross-sectional area, and the cross-sectional area occupied by the luminal mucosa was quantified as a ratio of the total cross-sectional area by using a computer image analysis system as reported by Hogg et al. 24. Digitised video images of the entire lung fields were analysed with a computerised colour image analysis software system (Win Roof Version 5.0; Mitani Co., Fukui, Japan) as recently reported elsewhere 20. Further details of these analyses are provided in the online supplementary data.
Immunohistochemical assay
Immunohistochemical analysis was performed as reported previously 15. Anti-human IL-18 (clone 8 (mouse immunoglobulin (Ig)G2a) and clone 1-8D (mouse IgG1)) monoclonal antibodies (mAbs) were used. IL-18 positive reactivity was identified by biotin-labelled goat anti-mouse and rabbit Ig, peroxidase-streptavidin and 3-3′-diaminobenzidine-4HCl (Dako, Kyoto, Japan). Double immunohistochemical analysis for IL-18 production in CD4-, CD8-, or CD13-positive cells was also performed. Anti-human CD4 (4B12 (mouse IgG2a); Novocastra, Newcastle upon Tyne, UK), anti-human CD8 (C8/144B (mouse IgG1); Dako) and anti-human CD13 (38C12 (mouse IgG1); Novocastra) mAbs were used. CD4, CD8 and CD13 positive reactivity was identified by the addition of biotin-labelled porcine anti-mouse, anti-rabbit and anti-goat Ig, alkaline phosphatase-streptavidin and Fast Red (Dako). Further details of these analyses are provided in the online supplementary data.
Quantitative assessment of alveolar macrophages and infiltrating mononuclear cells in the general interstitium of lung tissue
Quantitative assessment of alveolar macrophages and infiltration of mononuclear cells was performed as reported previously 15. Briefly, HE sections of lung tissues obtained from COPD patients who had undergone LVRS were viewed and, in each HE section, three square fields (2.5×3.5 mm2; 8.75 mm2), in which the small airway inflammation appeared most severe, were selected at 40× (fig. 1c⇓). Within each of these square fields, a further three individual square fields (1×1.4 mm2; 1.4 mm2) were selected at 100× (figs 2a⇓ and c). These smaller square fields were defined as the observation fields (OFs). Nine different OFs (9×1.4 mm2) were selected within three different square fields (3×8.75 mm2). The numbers of alveolar macrophages (AMs) and infiltrating mononuclear cells (IMNCs) in the general interstitium were counted within the nine OFs at 100× (figs 2a⇓ and c). For example, when 70 AMs were counted within one OF at 100×, the number of AMs was 50 cells·mm−2. The total number of AMs and IMNCs in nonsmokers, smokers and COPD patients was expressed as mean±sem cells·mm−2 (table 1⇓). Two pathologists examined these sections independently and in a blinded manner, without prior knowledge of the patients' clinical status.

Haemotoxylin and eosin staining of lung tissue samples from a) a non-smoker, b) a smoker and c) a chronic obstructive pulmonary disease patient. Scale bars = 500 μm.

Immunostaining of lung tissue samples with mouse anti-interleukin-18 monoclonal antibodies in two patients (a and b) patient 1; (c and d) patient 2; with Global Initiative for Chronic Obstructive Lung Disease stage IV chronic obstructive pulmonary disease. Scale bars = 100 μm (a and c) and 50 μm (b and d).
Interleukin(IL)-18 immunoreactivities in lungs of chronic obstructive pulmonary disease patients, smokers and nonsmokers
Quantitative assessment of cells producing IL-18
Percentages of IL-18 positive cells in AMs, IMNCs, CD4+, CD8+, and CD13+ cells were previously assessed as previously reported 15. Nine different OFs (9×1.4 mm2) were selected at 100× within three different square fields (3×8.75 mm2), as described previously. The OFs were scanned under a microscope at 400× (figs 2b⇑ and d). The percentage of cells that expressed IL-18 were counted within three different areas on each OF at 400×. Then, the mean percentage of cells that expressed IL-18 within nine OFs was calculated. The mean number of IL-18 positive cells·mm−2 in AMs and IMNCs (table 1⇑) was therefore calculated as:
(total no. of cells·mm−2)×(mean percentage of cells expressing IL-18)(1)
Two pathologists examined these sections independently, without prior knowledge of the patients' clinical status and in a blinded manner.
Serum levels of IL-18, IL-13, IL-4, and IFN-γ
Serum IL-18, -13, -4 and IFN-γ levels were measured with commercially available ELISA kits (supplied by Medical and Biological Laboratories Co. Nagoya, Japan for IL-18 and R&D Systems, Minneapolis, MN, USA for IL-13, IL-4, IFN-γ).
Statistical analysis
Results were expressed as means±sem. Nonparametric tests (Kruskal–Wallis and Mann–Whitney U-tests) were used to compare differences between the groups. Correlations were analysed by simple regression. A p<0.05 was considered to be statistically significant.
RESULTS
Clinical findings
Tables 2⇓ and 3⇓ provide details (number, age, sex, GOLD stage, smoking history, body mass index, pulmonary function and treatment) of all individuals subjected to the immunohistochemical and ELISA analyses. All of the 16 COPD patients who had undergone LVRS had stopped smoking 2–21 yrs previously (mean 9.5±1.8 yrs). Three COPD patients who had undergone LVRS had received inhaled beclomethasone dipropionate 400 μg·day−1 and three were receiving 800 μg·day−1. All 16 patients were receiving bronchodilators, such as β2-agonists, anticholinergics and/or methylxanthines. Sera were obtained from 62 COPD patients, 47 nonsmokers and 34 current smokers for ELISA analysis. In total, 30 of these COPD patients had stopped smoking 2–21 yrs previously (mean 7.6±1.0 yrs). Four patients with severe COPD (GOLD stage III) were receiving inhaled fluticasone 400 μg·day−1, and one was receiving 800 μg·day−1. Five patients with very severe COPD (GOLD stage IV) were receiving fluticasone 400 μg·day−1 and five were receiving 800 μg·day−1. All of the 15 COPD patients receiving fluticasone also received bronchodilators, such as β2-agonists, anticholinergics and/or methylxanthines. All the COPD patients analysed were clinically stable and experienced no disease exacerbations during the previous 3 months.
Clinical characteristics of chronic obstructive pulmonary disease(COPD) patients, smokers and nonsmokers examined by immunohistochemical analysis
Clinical characteristics of chronic obstructive pulmonary disease(COPD) patients, smokers and nonsmokers examined by ELISA analysis
Persistent and severe airway inflammation in COPD patients
Severe small airway remodelling, such as increase in the thickness of the wall, hyperplasia of the mucosa and presence of mucous exudates in the lumen, was observed in the lungs of COPD patients (fig. 1c⇑). The computer image analysis system previously reported 24 was used to quantify both the wall area and the luminal mucosa area as ratios of the total airway area in six COPD stage IV patients who had undergone LVRS, six smokers, and six nonsmokers. The wall to total area ratios were 34.7±2.5% (COPD patients), 8.8±0.9% (smokers), and 10.3±1.1% (nonsmokers); and the respective luminal mucosa to total area ratios were 18.7±1.5%, 11.2±0.9%, and 9.5±1.2%, respectively. Both ratios in COPD patients were significantly (p<0.01) higher than those in smokers and nonsmokers. Computer image analysis clearly revealed severe airway remodelling in the COPD patients, as reported by a previous study 24. Quantitative analysis revealed that the numbers of AMs and IMNCs in the lungs of patients with very severe COPD who had stopped smoking >2 yrs previously were significantly (p<0.05) greater than in nonsmokers and smokers. The numbers of AMs and IMNCs were also significantly (p<0.05) greater in the lungs of smokers than of nonsmokers. There were no significant differences in the numbers of AMs and IMNCs in the lungs of male and female smokers (16 versus 11, respectively) or in the lungs of male and female nonsmokers (10 versus 13, respectively; table 1⇑).
Increased expression levels of IL-18 in the lungs of COPD patients
The airway epithelial cells in the lungs of nonsmokers weakly expressed IL-18 protein, as previously reported 15. Approximately 30–50% of AMs in the lungs of nonsmokers and smokers weakly or modestly expressed IL-18 protein (table 1⇑). In contrast, IL-18 protein was strongly expressed in ∼80% of AMs and in some IMNCs in the lungs of COPD patients (fig. 2⇑). In addition, IL-18 was strongly expressed in both the bronchiolar and alveolar epithelium in patients with very severe COPD (GOLD stage IV; fig. 3⇓). Microscopic tissue examination showed that type-2 pneumocytes expressed mainly IL-18 proteins. Quantitative analysis showed that IL-18 expression levels were significantly (p<0.05) greater in AMs and IMNCs in the lungs of COPD patients than in those of nonsmokers and smokers. There were no significant differences between males and females in terms of IL-18 expression levels in AMs and IMNCs in the lungs of nonsmokers or smokers (table 1⇑). Excess recombinant human IL-18 (10 μg·mL−1) completely blocked all positive reactivity with anti-human-IL-18 antibodies (data not shown). These results indicated that anti-human-IL-18 mAbs specifically recognised IL-18 expression in the lungs of COPD patients.

Immunostaining of lung tissue samples with mouse anti-interleukin-18 monoclonal antibodies in chronic obstructive pulmonary disease patient 2. Scale bars = 50 μm.
Numbers of IL-18-producing CD8+ T-cells were increased in the lungs of COPD patients
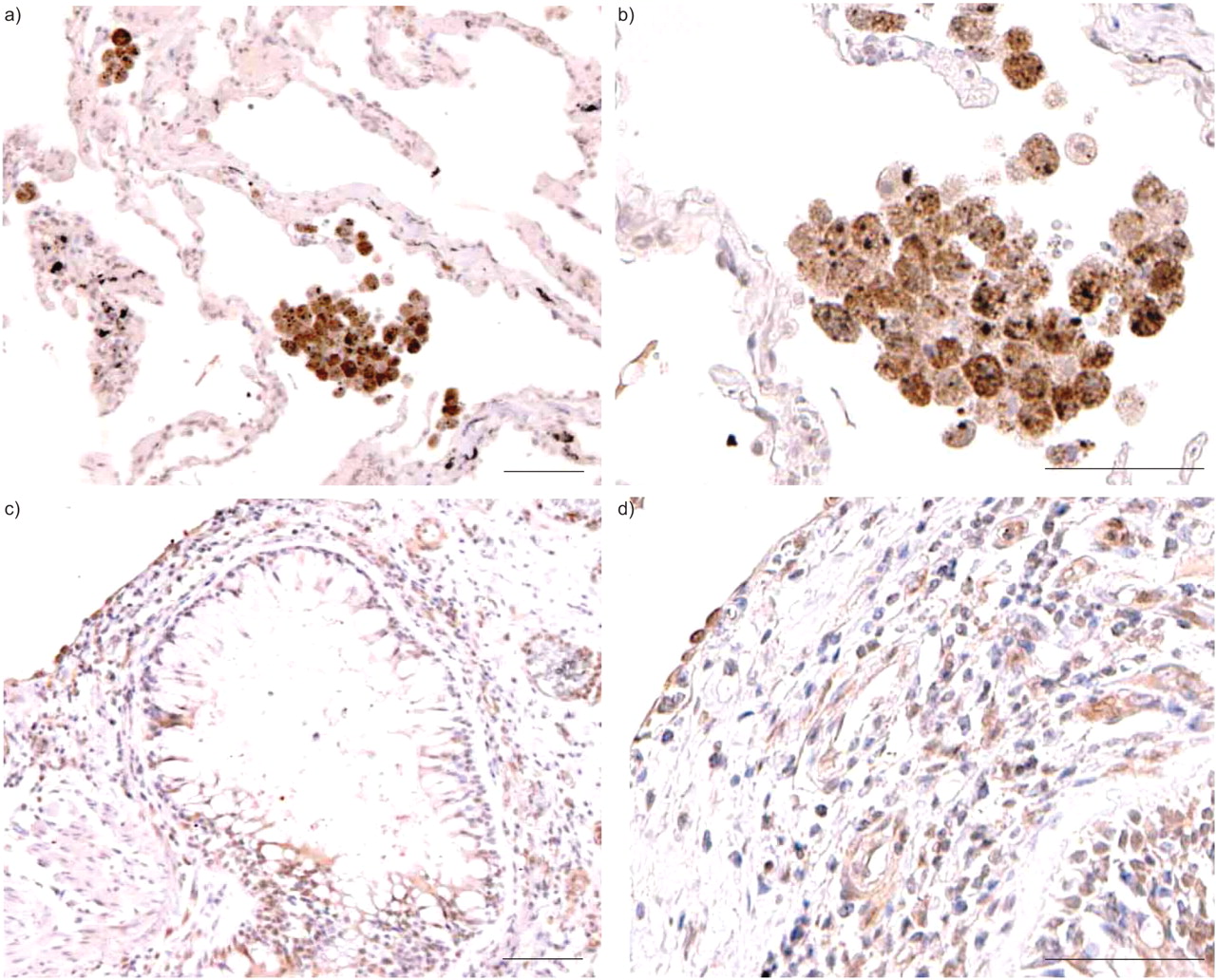
Double immunostaining was performed with two different antibodies on one lung tissue slide in order to identify whether CD8+, CD4+, or CD13+ T-cells (presumably monocytes, eosinophils or macrophages) among the IMNCs produced IL-18 proteins in 16 patients with very severe COPD. CD8, CD4 and CD13 positive cells were identified by Fast Red using fluorescence microscopy. Approximately 50–80%, 10–20% and <10% of the IMNCs in the lungs of the 16 patients were CD8+, CD4+, and CD13+ cells, respectively (fig. 4⇓, and data not shown). Of these, 91.5, 87.1 and 94.6%, respectively, produced IL-18. The percentages of IL-18-positive CD8+, CD4+, and CD13+ cells did not differ significantly from each other (fig. 5⇓). IL-18–producing CD8+ T-cells were barely observed in the lungs of nonsmokers and smokers (data not shown). These results showed that the proportions of IL-18-producing CD8+ T-cells were increased in the lungs of patients with very severe COPD.

Double immunostaining of lung tissue samples with a) mouse anti-CD8 alone and b) in combination with mouse anti-interleukin (IL)-18 monoclonal antibodies on one slide in chronic obstructive pulmonary disease patient 2. CD8 positive cells were identified with Fast Red staining under a fluorescent microscope (arrows). IL-18 positive reactivity was identified by 3-3′-diaminobenzidine-4HCl treatment (b). Scale bars = 50 μm.
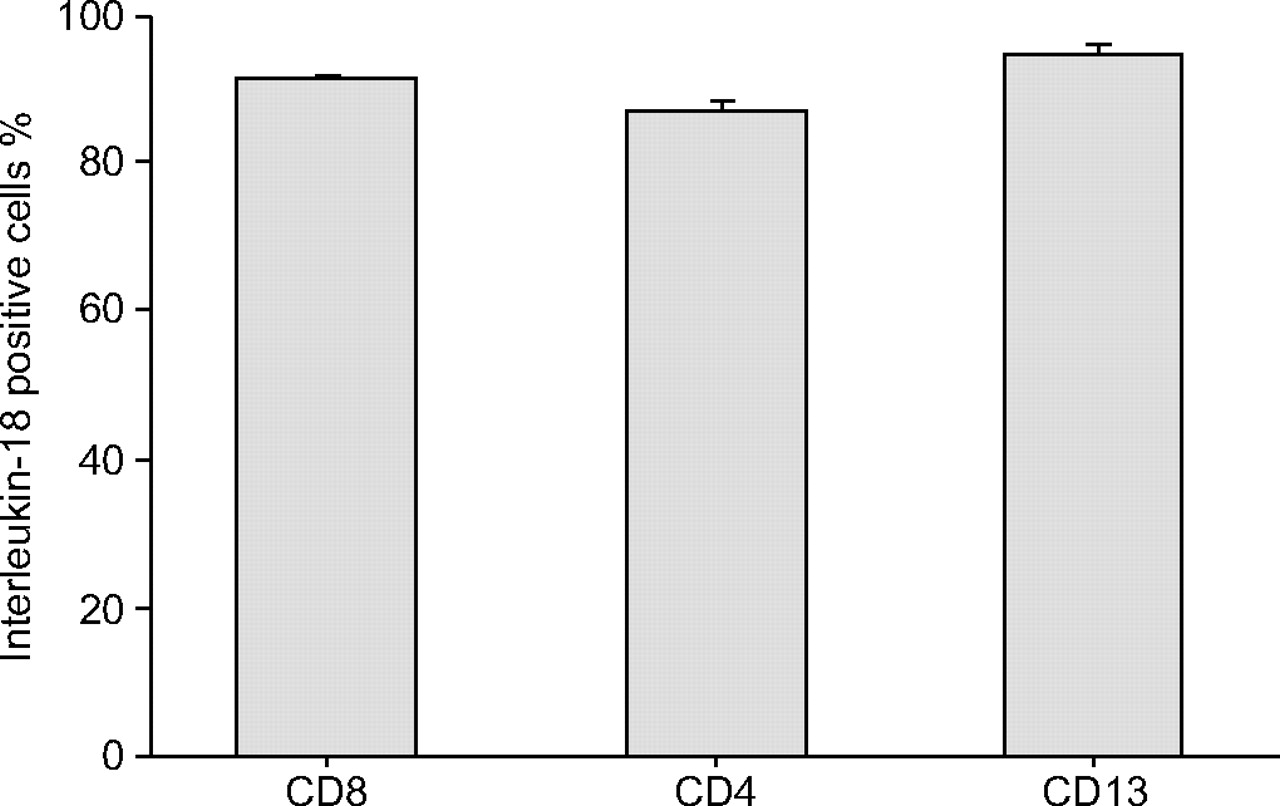
Interleukin-18 positive cells in CD8+, CD4+ and CD13+ cells in the lungs of 16 chronic obstructive pulmonary disease patients. Whiskers represent sem.
Increase in serum levels of IL-18 but not IL-13, -4, or IFN-γ in COPD patients
The immunohistochemical analysis suggested that IL-18 proteins produced in the lungs were circulating in the sera of COPD patients. Therefore, serum levels of IL-18, -13, -4, and IFN-γ were analysed in COPD patients, smokers and nonsmokers. Serum levels of IL-18 in COPD patients (n = 62) and smokers (n = 34) were significantly (p<0.05) higher than in nonsmokers (n = 47; table 4⇓). Next, the 62 COPD patients were categorised according to the GOLD classification of severity of COPD 22. Serum IL-18 levels in GOLD stage I (n = 14), II (n = 19), III (n = 16) and IV (n = 13) COPD patients were 178.2±18.3, 185.7±9.8, 267.0±23.4, and 293.9±19.3 pg·mL−1, respectively. Serum levels of IL-18 in GOLD stages III and IV were significantly (p<0.05) higher than those in smokers and nonsmokers (fig. 6⇓). There were no significant differences in serum IL-18 levels between current and ex-smokers within the COPD group. Smoking history in patients with COPD stages I, II, III and IV were 53.4±8.3, 60.0±7.1, 53.3±5.8 and 50.0±4.0 pack-yrs, respectively. There were no significant correlations between smoking history (pack-yrs) and serum levels of IL-18 in smokers and COPD patients. There were no significant differences in serum IL-18 levels between males and females in the nonsmoker, smoker or COPD groups (data not shown). Serum levels of IL-13, -4, and IFN-γ were not significantly increased in smokers or in COPD patients (table 4⇓).

Serum levels of interleukin-18 in nonsmokers (n = 47), smokers (n = 34) and chronic obstructive pulmonary disease (COPD) patients classified according to Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages I (n = 14), II (n = 19), III (n = 16) and IV (n = 13). *: p<0.05 versus nonsmokers; ¶: p<0.05 versus smokers; +: p<0.05 versus patients with GOLD stage I COPD and p<0.05 versus patients with GOLD stage II COPD.
Serum levels of interleukin(IL)-18, -13, -4 and interferon (IFN)-γ in chronic obstructive pulmonary disease (COPD) patients, smokers and nonsmokers
Negative correlation between serum levels of IL-18 and FEV1% pred
The correlation between serum levels of IL-18 and pulmonary function was analysed in nonsmokers, smokers and COPD patients. In COPD patients, there was a significant (p<0.001) negative correlation between serum level of IL-18 and FEV1 % pred but not between serum IL-18 and FEV1/FVC % (r = 0.4844; fig. 7⇓). In contrast, no significant correlations between serum levels of IL-18 and FEV1 % pred were observed in nonsmokers or smokers.
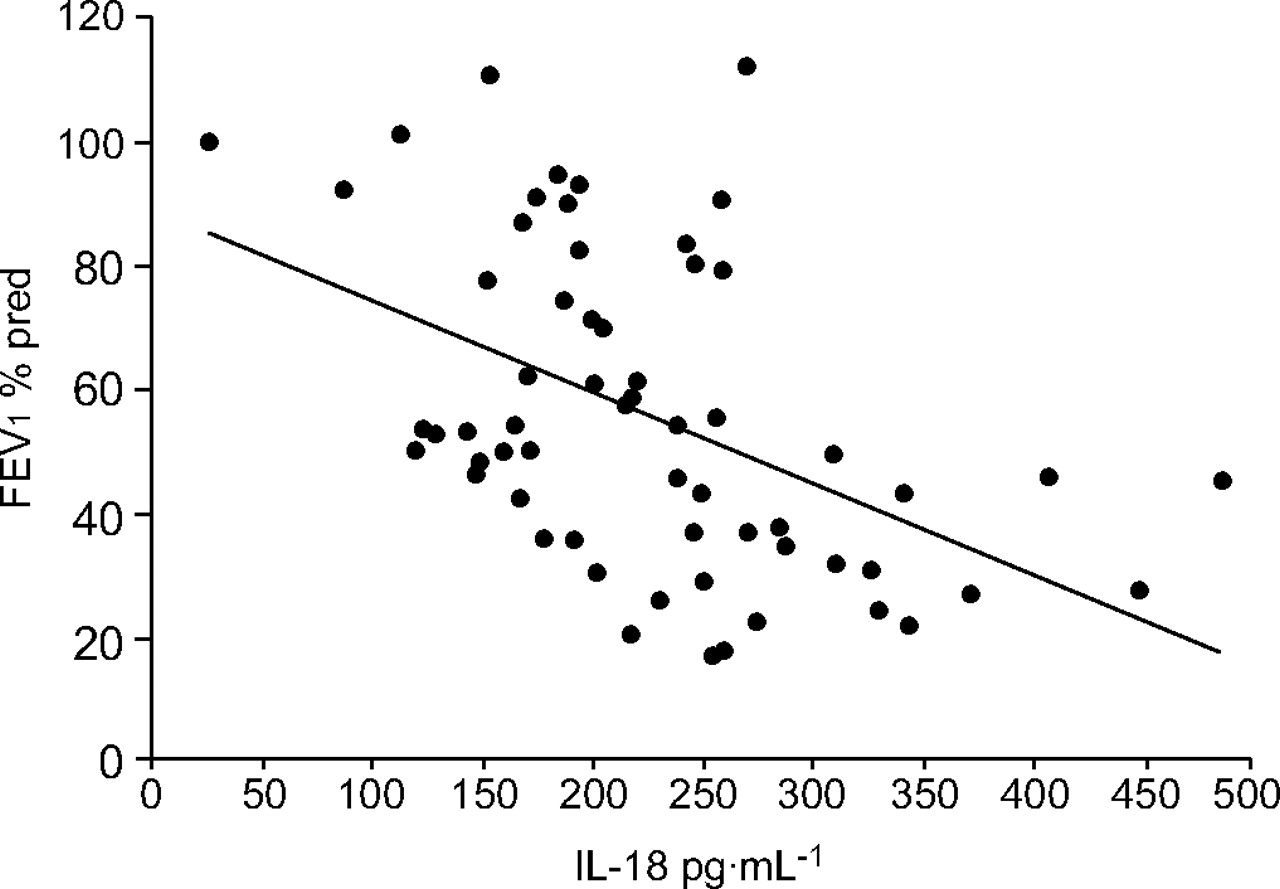
Correlation between serum levels of mature interleukin (IL)-18 and lung function measured as forced expiratory volume in one second (FEV1) % predicted in chronic obstructive pulmonary disease patients (n = 62). The gradient and intercept of the best-fit line are -0.14721 and 88.9083, respectively (r = 0.4844 and p<0.001).
DISCUSSION
In the present study, it was shown that IL-18 was strongly expressed in AMs and CD8+ T-cells in the lungs of patients with very severe COPD. The enhanced IL-18 protein production in the AMs and CD8+ T-cells of COPD patients was presumably of both the pro- and mature IL-18 forms, as the anti-human-IL-18 monoclonal antibody (clone 8) used in the study recognises both forms 15. However, when an ELISA specific for the mature form of IL-18 was used 15, serum levels of mature IL-18 protein in COPD and smokers were found to be significantly higher than in nonsmokers. A recent paper reported that cigarette smoke induced IL-18 production in the lungs of mice and that serum levels of IL-18 were increased in COPD patients 25. The present authors found that: 1) IL-18 protein was strongly expressed in the majority of AMs and CD8+ T-cells, and in both the bronchiolar and alveolar epithelia of COPD patients; 2) the levels of mature IL-18 protein were significantly greater in the sera of patients with GOLD stage III and IV COPD than in nonsmokers or smokers; and 3) a significant correlation existed between serum level of IL-18 and pulmonary function (FEV1 % pred) in COPD patients, but not in non-smokers or smokers. These results suggest that cigarette smoke induces the production of large amounts of reactive oxygen species, activates the cascade of cathepsin-B–caspase-11–caspase-11, and induces mature IL-18 production 26 in AMs and IMNCs in the lungs of COPD patients, leading to higher levels of mature IL-18 in the sera of these patients. Previous studies have shown that mature IL-18 proteins are tightly bound to IL-18 binding protein 27 and/or IgM 28 in human sera. Mature IL-18 complexed with IL-18BP and/or IgM does not degrade readily, and this complex is easily detectable by ELISA. It has been reported that FEV1 % pred is an important prognostic factor in COPD 29, 30. The present study showed that a significant correlation existed between serum level of IL-18 and pulmonary function (FEV1 % pred) in COPD patients. Therefore, mature IL-18 in the sera can be used as a new biomarker for disease activity in COPD. However, the mechanisms controlling cytokine production usually differ between lung tissue and peripheral blood. Analysis of IL-18 protein expression in lung and blood samples from the same COPD patients may be necessary to verify this issue. However, the present authors were unable to obtain paired tissue and blood samples from COPD patients. Further analysis is needed to confirm this model.
Previous studies have shown that there is persistent inflammation in the small airways of both ex-smokers and smokers. For instance, airway obstruction and chronic expectoration are associated with increased numbers of neutrophils in the sputum of smokers and ex-smokers 31. Induced sputum samples obtained from COPD patients who have stopped smoking and from current smokers with COPD show increased neutrophil numbers 32. The presence of persistent and severe small airway inflammation has been confirmed in the resected lung tissues of ex-smokers with very severe COPD. The present results show that cessation of smoking cannot prevent pulmonary inflammation in severe COPD.
It has been reported that oestrogen levels can modulate the production of some cytokines from peripheral blood mononuclear cells 33, suggesting that sex differences may influence the production of IL-18. Therefore, the effect of sex on numbers of AMs and IMNCs and on IL-18 expression levels was analysed in the lung tissues obtained from nonsmokers and smokers. In nonsmokers or smokers there were no significant differences between males and females in terms of the numbers of AMs and IMNCs or IL-18 expression levels. Moreover, levels of IL-18 in the sera were not significantly different between males and females in the nonsmoking, smoking or COPD groups (data not shown). Although lung tissues from female patients with very severe COPD could not be obtained, the present results suggest that sex differences may not influence IL-18 production.
The numbers of AMs and IMNCs were significantly greater in smokers than in nonsmokers. These results suggest that acute smoking may induce pulmonary inflammation in both smokers with COPD and smokers with normal lung function. The present authors wanted to compare the levels of IL-18 protein expression in the lungs of current smokers with normal lung function and of current smokers with very severe COPD (GOLD stage IV). However, lung tissues from patients with very severe COPD who were current smokers could not be obtained. Further analysis is needed to verify this issue.
Six patients with very severe COPD (GOLD stage IV) were receiving inhaled corticosteroids (ICS) at the time of LVRS. A total of 15 patients with COPD (GOLD stage III and IV) were using ICS when their sera were obtained for ELISA analysis. None of the COPD patients analysed had been receiving systemic steroid therapy. It is of note that there were no significant differences between COPD patients treated or not treated with ICS in terms of IL-18 expression levels in the lungs and sera (data not shown). These results suggest that IL-18 production may not be influenced by ICS treatment. However, a previous study 34 reported that steroids suppressed the production of IL-18 in lipopolysaccharide-/IL-2-stimulated peripheral blood mononuclear cells in vitro. Further analysis is needed to verify whether steroids can influence IL-18 production in COPD patients.
As described previously, IL-13 and IFN-γ are thought to play important roles in the pathogenesis of COPD 7, 12, 13. It has been previously demonstrated that IL-18 induces the production of both Th1- and Th2-type cytokines, including IL-13, -4 and IFN-γ, both in vitro and in vivo 16–18. Very recently, the present authors established lung-specific IL-18 Tg mice that constitutively overproduce IL-18 in their lungs. Constitutive overproduction of IL-18 in the lungs results in: increased production of both Th1 and Th2 cytokines, including IFN-γ and IL-13; emphysematous changes; and severe pulmonary inflammation in mice 20. Increased numbers of CD8+ T-cells are characteristic pathological features of the lungs in COPD 3, 4. In the present study, it was shown that the numbers of CD8+ T-cells were increased in the lungs of patients with very severe COPD, and ∼90% of these CD8+ T-cells produced IL-18 proteins. The majority of CD8+ T-cells and alveolar macrophages in the lungs of COPD produced IL-18. Enhanced IL-18 production may induce the subsequent localised production of Th1- or Th2-associated cytokines in the lungs of COPD patients. IL-18-producing CD8+ T-cells and alveolar macrophages may be involved in the pathogenesis of COPD. Further analysis is needed to confirm this hypothesis.
The treatment strategy for chronic obstructive pulmonary disease consists mainly of the use of bronchodilators, such as β2-agonists, theophylline and anticholinergics 22. There is no effective therapy to reduce the persistent pulmonary inflammation in chronic obstructive pulmonary disease patients and improve their prognosis, even in those patients who use inhaled corticosteroids 35, 36. Therefore, the disease is being targeted with new anti-inflammatory treatments. In the present study, the overproduction of interleukin-18 in the lungs of chronic obstructive pulmonary disease patients has been demonstrated. The present results raise the possibility that blockade of interleukin-18 may be a feasible treatment for chronic obstructive pulmonary disease. Caspase-1 inhibitors, antibodies to interleukin-18 and their receptor, interleukin-18 binding protien, or inhibitors of genes downstream of the interleukin-18 signal transduction pathway, such as those encoding MyD88, interleukin-1 receptor associated kinase, tumour necrosis factor receptor-associated factor 6, nuclear factor-κB, C-jun N-terminal kinase, and p38 mitogen-activated protein kinase, as well as interleukin-13 inhibitors, may be of clinical benefit in the treatment of severe chronic obstructive pulmonary disease patients who have poor clinical prognoses.
Support statement
This work was supported by: a Grantin-Aid for Scientific Research (B); (no. 17390244) and a Grant-in-Aid for Exploratory Research (no. 18659244) from the Ministry of Education, Science, Sports and Culture of Japan; a grant from the Kakihara Foundation (Fukuoka, Japan); a grant from the Takeda Science Foundation (Osaka, Japan) to T. Hoshino; and a Grant-in-Aid for Scientific Research (B); (no. 18390244) to H. Aizawa.
Statement of interest
None declared.
Acknowledgments
The authors would like to thank T. Iwanaga for contributing to the study. The authors also thank: H. A. Young (NCI-Frederick) for editing; K. Ohshima for helpful discussions; and T. Koga, Y. Kitasato, Y. Sakazaki, K. Azuma, R. Toda, N. Edakuni (Kurume University), T. Shirakusa (Fukuoka University), Y. Doi (Chikugogawa Onsen Hospital), T. Tsuda (Kirigaoka Tsuda Hospital), T. Shigemoto (Shigemoto Hospital), M. Kawahara, H. Koga (Keisinkai Hospital), N. Tokunaga (Tokunaga-Naika Clinic), N. Hachiya (Social Insurance Futase Hospital) and T. Kukita (Arao Central Hospital) for supplying the COPD tissue samples. The authors would also thank E. Kuma, C. Harada, E. Hidaka and K. Yamaguchi for their technical assistance.
Footnotes
-
This article has supplementary material accessible from www.erj.ersjournals.com
- Received February 16, 2007.
- Accepted October 24, 2007.
- © ERS Journals Ltd











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}