





Abstract
Asthma and chronic obstructive pulmonary disease (COPD) are characterised by airflow obstruction, airway remodelling (measurable structural change) and inflammation. The present review will examine the relationship between airway remodelling in these two conditions with respect to symptoms, abnormal lung function, airway hyperresponsiveness and decline in lung function. The potential for remodelling to be a protective response will also be discussed.
Asthma is associated with variable symptoms and changes in lung function and also fixed abnormalities of lung function and an increased rate of decline in lung function with age. There is a relative preservation of the relaxed airway lumen dimensions, prominent thickening of the smooth muscle layer and reduced airway distensibility. The severity of asthma is related to the degree of airway remodelling, which is most marked in cases of fatal asthma.
In COPD, symptoms are persistent and predictable but also progressive and are related to fixed abnormalities of lung function. Remodelling is associated with narrowing of the airway lumen and an increased thickness of the airway wall, although not usually to the extent seen in asthma. COPD is most often due to smoking where there is also remodelling of the parenchyma that may contribute to symptoms.
SERIES “AIRWAY REMODELLING: FROM BASIC SCIENCE TO CLINICAL PRACTICE”
Edited by L-P. Boulet and P.J. Sterk
Number 4 in this Series
The specific elements of remodelling which contribute to the clinical and functional manifestations of asthma and chronic obstructive pulmonary disease (COPD) remain poorly understood, due to the heterogeneity of these “diseases” and the interactions of numerous structural components within the airways. This article will examine the evidence that airway remodelling influences the clinical expression and natural evolution of airway diseases, predominantly asthma and COPD, and assess the relative protective and detrimental effects of this process. Previous articles in this series have defined remodelling and its relationship to airway function. This article will attempt to link what is known about airway remodelling to the acute and chronic clinical expression of airway disease, including symptoms, exacerbations, progression and severity of disease. This will not include the effects of remodelling in the integrated airway tree, which is suggested by the overall reduction of airway complexity seen on casts from cases of fatal asthma 1 and by functional imaging studies demonstrating altered distribution of airflow in cases of asthma during bronchoconstriction with varying airway closure 2, 3.
Asthma is defined as “a chronic inflammatory disorder of the airways in which cells and cellular elements play a role. The chronic inflammation is associated with airway hyperresponsiveness that leads to recurrent episodes of wheezing, breathlessness, chest tightness and coughing, particularly at night or in the morning. These episodes are usually associated with widespread but variable airflow obstruction that is often reversible either spontaneously or with treatment” 4. This less-than-perfect operative definition relates symptoms to airway hyperresponsiveness (AHR) and airway inflammation. In asthma, the pathology seems confined primarily to the conducting airways. Not only is airway inflammation present but there is also considerable structural remodelling of the airway wall, particularly the smooth muscle. The airway hyperresponsiveness that is characteristic of asthma is independent of baseline lung function. Symptoms and abnormalities of lung function generally (but not always) respond well to treatment with bronchodilators and inhaled or oral corticosteroids, but recur if treatment is withdrawn.
COPD is defined by the presence of persistently abnormal lung function, most often measured using forced expired volume in one second (FEV1) 5–7. The pathology of COPD involves both the conducting airways and the lung parenchyma, and COPD is associated with a systemic component 8. It is characterised clinically by varying severity of cough and sputum production and by persistent, predictable breathlessness related to exertion. Symptoms and lung function deteriorate with time, especially in those who continue to smoke. Unlike in asthma, AHR in COPD is generally related to airway calibre and the disease responds poorly to treatment with bronchodilators or corticosteroids 9. In a similar manner to asthma, exacerbations in COPD are frequently caused by viral infections 10. COPD is most often, but not always, caused by smoking cigarettes. However, it should be remembered that some patients with asthma (who may or may not smoke cigarettes) may develop fixed or irreversible (by treatment) airflow obstruction and thus, by definition, also have COPD.
Remodelling and inflammation of the airways are characteristic of both asthma and COPD. The present article will regard remodelling as a quantifiable change in the dimensions of airway structure or in the relationship of airway structures to each other, compared with normal individuals. Remodelling in asthma and COPD and how this may be related to clinical outcomes will be separately examined, recognising that many of the clinical and pathological features of these two conditions overlap.
Although uncommon, asthma continues to cause death in some patients. Fatal asthma usually occurs on a background of chronic, severe symptoms often with fixed abnormalities of lung function, recurrent admissions to hospital, poor use of treatment or poor response to it, psychological factors and an underestimation of disease severity. A large body of qualitative and quantitative observations of airway structure in fatal asthma exists, and suggests that airway remodelling has a crucial role to play in fatal attacks. Much of the data regarding asthma pathology, especially remodelling, has been obtained from cases of fatal asthma. The extent to which these cases represent a uniquely remodelled phenotype or reflect clinically severe asthma will also be discussed, although this remains poorly understood.
Finally, it is largely assumed that airway remodelling is detrimental. There is plenty of evidence to support this view and this article will add to this impression. However, there is a growing interest in the possible protective role of remodelling which may be thought of as either adaptive remodelling that is designed to protect, yet in fact is detrimental, or remodelling that is truly protective and without which the patient would be even more severely compromised by symptoms and abnormal lung function.
REMODELLING: THE CHANGES IN ASTHMA AND COPD
Epithelium
Although desquamation of the epithelium is often reported as a pathological feature of asthma 11–15, quantification of the amount of damaged or missing epithelium in cases of mild or severe asthma has not shown any systematic significant differences from nonasthmatic control cases 16–18. This is not surprising since damage to the epithelium in bronchial biopsies or as a post mortem artefact is considerable and likely to confound structural differences between cases of asthma and normal controls. What is clear is that in asthma the epithelium is thicker 17, there is hyperplasia of goblet cells (fig. 1⇓) 19, 20 and there is increased turnover of epithelial cells, suggested by the more frequent appearance of free epithelial cells in bronchoalveolar lavage fluid 21, 22 or sputum 23, or the reduced viability of epithelial cells 24 in patients with asthma. These findings are not universal: some studies have shown no differences in the number of epithelial cells in the lavage of asthmatic patients 25 and it is apparent that significant desquamation of epithelial cells can occur in biopsy specimens from nonasthmatic subjects 26.

Section of airway epithelium from an asthma patient showing marked increase in goblet cell size and number and deposition of matrix material below the basement membrane. Periodic acid-Schiff stain. Scale bar = 25 μm.
In COPD, epithelial thickness is also increased 27 and shows goblet cell hyperplasia 28, 29. In addition, metaplasia of the epithelium is a more characteristic pathological change 30, compared with normal subjects or patients with asthma, although this is more an effect of cigarette smoking than the development of COPD.
The epithelium in asthma and COPD is a site of inflammation, displays altered permeability and is the source of a number of growth factors and cytokines which modulate inflammation and remodelling below the basement membrane 31. These aspects of epithelial pathology are beyond the scope of this article. The role of epithelial remodelling in the clinical manifestations of airway disease is discussed below.
Smooth muscle
The increased thickness of the smooth muscle contributes most to the increased airway wall thickness observed in asthma 32. Increased thickness of the smooth muscle layer seen on transverse airway sections could result from more muscle cells (hyperplasia), larger muscle cells (hypertrophy) and/or more extracellular matrix (ECM) within the smooth muscle layer. A few studies have suggested that hyperplasia of smooth muscle is present in the central airways in some fatal cases 33, 34 and in clinically mild-to-moderate cases 35 of asthma. The latter study did not find any hypertrophy of smooth muscle or changes in contractile proteins that might suggest a hypercontractile smooth muscle cell phenotype. Conversely, Ebina and co-workers 33, 36 showed that in cases of fatal asthma where the smooth muscle was increased in both the central and peripheral airways, hyperplasia and hypertrophy were present in the central airways and hypertrophy was also present in the peripheral airways. These carefully conducted studies, undertaken using stereological methods, did not take into account the effects of the ECM between smooth muscle cells. This may differ systematically between cases of asthma and normal subjects, and estimation of the volume fraction of ECM may depend critically on the orientation of the smooth muscle layer that is used (fig. 2⇓) 37. For example, an increase in ECM in the smooth muscle layer may lead to an overestimation of smooth muscle volume and the degree of hypertrophy, since mean individual cell volume in tissues is calculated by dividing the number of cells (nuclei) into the estimated smooth muscle volume.

Airway smooth muscle stained with the Masson's trichrome technique and cut in: a) longitudinal section (4 μm) showing extensive overlap of the smooth muscle (red) and extracellular matrix (blue); b) transverse section showing separation of the smooth muscle and matrix; and c) thin longitudinal section (0.5 μm) also showing separation of muscle from the matrix. These images emphasise that changes in the amount and interaction of the matrix and smooth muscle in airway diseases are likely to affect airway function. Scale bars = 50 μm.
An increase in the volume of smooth muscle does not necessarily mean increased force development, since proliferating or secretory smooth muscle may have reduced contractility 38, 39. A hypercontractile phenotype is observed in vitro after serum deprivation or other manipulations of cultured smooth muscle cells 39. It is not clear whether the “normal” state of smooth muscle in situ is secretory, contractile or hypercontractile. The smooth muscle from patients with asthma has not been shown to contain more contractile proteins 35, but it is more sensitive to proliferative stimuli 40. Theoretical studies have suggested that the smooth muscle in the thickened asthmatic airway would probably have to generate more force to narrow the thickened and (theoretically) less compliant airway 41, although biological confirmation of these calculations does not yet exist.
Airway smooth muscle (ASM) has been assessed in a number of studies of COPD. Hogg et al. 42, in their landmark paper in 1968, showed with semi-quantitative pathology that smooth muscle was increased in the small airways in patients who had more severe airflow obstruction, were older and had smoked more cigarettes. Subsequent studies of smooth muscle in COPD have shown mixed results, with approximately half showing either an increase 27, 43–45 or no significant change 46–48. In studies that also included comparisons with cases of asthma, the area of smooth muscle was less in COPD than in asthma 45, 49. The study of Tiddens et al. 48 was one of few to study large airways in patients who had been harmed by cigarette smoke and had developed airflow obstruction. However, unlike that of Dunnill et al. 49, the former study found no significant differences in the amount of ASM in the large (or small) airways. In addition, it was found that, although maximum expiratory flow rates were related to overall wall area, they were not related to the area of ASM.
In summary, many studies show that there is more ASM in cases of asthma and COPD than in control cases, but there are some studies that do not agree and others that show heterogeneity between cases and between small and large airways. The contribution of the ECM to the smooth muscle layer in asthma and in COPD also remains to be determined.
ECM
Increased attention has recently been directed to the tissue between (and within) the structures of the airway wall: the ECM. This amorphous material is made up of fluids and a combination of proteoglycans and glycosaminoglycans. The ECM proteins have a number of functions that include: structural integrity; fluid balance; cellular migration; assembly and aggregation of structural proteins; elaboration of growth factors and cytokines; and osmotic activity. Changes in the relative amounts of these matrix proteins may therefore lead to changes in function. The increase in ECM between ASM cells has been suggested to cause the increased area of ASM observed in asthma (fig. 2⇑) 37; it may also alter airway wall compliance 50. Studies of matrix proteins in asthma have shown increased deposition of: collagen, particularly sub-types I, III and IV 51–54; decorin 55, 56; laminin 57, 58; tenascin 53, 59, 60; versican 55, 56, 59; biglycan 56, 59, 61; perlecan 56, 61; hyaluranon 50, 62; and decreased lumican 55. Reductions in elastin have been observed in a number of studies 63–65. Increased collagen in the airway submucosa has also been observed in occupational asthma 66. Recently, researchers have begun to examine the ECM within the bundles of ASM, although the results are only currently available in abstract form. These show that elastin 67 is increased in the smooth muscle layer in large airways in cases of fatal asthma, compared with control cases. No significant differences in total collagen content were observed 68. Pini et al. 69 showed that, while levels of proteoglycans were unchanged in the subepithelial layer, they were increased in the smooth muscle layer in moderate asthma, compared with severe cases of asthma.
Fewer studies have been undertaken in COPD but a number have reported “fibrosis” in the airway wall of smokers with COPD 43, 46, 70. Interestingly, there are no studies that have shown changes in airway wall collagen in COPD. Increased deposition of tenascin and laminin has been reported in asymptomatic smokers 71 and has been related to the numbers of mast cells in the airway wall 72. Van Straaten et al. 73 showed that airways from smokers with severe emphysema had diminished staining in peribronchial tissues for the proteoglycans decorin and biglycans, without changes in collagen, fibronectin or laminin, compared with nonsmokers or those with mild emphysema.
Remodelling of other structures in asthma and COPD
A number of studies have examined the vasculature of the airway wall in asthma and have shown an increase in the number 74 or area 75, 76 of the submucosal small blood vessels. These changes are thought to be due to both dilatation and congestion of existing airways but also to the formation of new vessels, possibly in response to local inflammation and the elaboration of growth factors 77. Remodelling of large peribronchial vessels has also been observed 78, along with thickening of the arterial media in cases of fatal asthma. Kuwano et al. 45 found no significant differences between blood vessel dimensions in cases of COPD and control cases. The effects of the reported changes in blood vessels in asthma on clinical manifestations may be many. First, congested vessels will occupy space and may contribute to the exaggerated airway narrowing that occurs in response to smooth muscle shortening around a thickened airway 79, 80. However, this effect is likely to be small given the extent of the changes observed and the absence of large vessels between the ASM and airway lumen 76. Secondly, dilatation of vessels or rapid movement of fluid from vessels into peribronchial tissues may dissociate the elastic load of the lung parenchyma from contracting smooth muscle 81. Thirdly, exudation of fluid may also contribute to increased thickness of the inner airway wall. Fourthly, it has been shown that reduced bronchial blood flow may prolong and accentuate airway responses to bronchoconstricting stimuli, probably by delaying the removal of an inhaled stimulus 82. The extent to which these effects operate in asthma or COPD is unknown.
Quantitative studies of the local innervation of the airways in asthma have shown conflicting responses regarding vaso-active intestinal peptide-positive nerves 83, 84. Substance P-immunoreactive nerves were increased in cases of cough without any difference between cases of asthma and normal controls 85. There are no published quantitative studies of nerve tissues in COPD, although it has been suggested that there is more evidence for disrupted neural control in asthma than in COPD 86.
The cartilage may act as a significant load, which limits shortening of the ASM, because its removal or a reduction in its stiffness results in excessive airway narrowing following stimulation of the ASM 87, 88. The amount of cartilage has been shown to be increased or unchanged in asthma 32, and one study showed reduced cartilage in COPD 89.
EVIDENCE THAT REMODELLING INFLUENCES THE CLINICAL EXPRESSION OF AIRWAY DISEASES
Symptoms
The diagnosis of asthma is based largely on a characteristic history of intermittent symptoms, predominantly wheezing, chest tightness, shortness of breath, cough and mucus production. The variability of these symptoms, their precipitation by factors such as exercise, cold air, allergens, viral respiratory infections or specific agents such as aspirin, their relief by bronchodilators and control by inhaled corticosteroids, form the basis of the clinical diagnosis of asthma. Doctor-diagnosed asthma is the current gold standard for diagnosing asthma in most population studies. It is considered that wheeze, chest tightness and breathlessness (and possibly cough) result from excessive narrowing of large and small airways and subsequent air-trapping in asthma. Although COPD is defined by abnormalities of lung function, the recent Global Initiative for Chronic Obstructive Lung Diseases (GOLD) classification 5 recognises that chronic bronchitis (daily cough and sputum production) may be present with or without airflow obstruction. The symptoms of COPD are in part related to the remodelling of airways and in part related to changes in parenchymal structure and function. The latter include reduced elastic recoil, reduced alveolar surface area and ventilation–perfusion inequality due to emphysema, altered chest wall mechanics due to chronic hyperinflation, and systemic problems such as muscle wasting in severe cases. However, it is clear that in COPD narrowing of the airway lumen also contributes to symptoms.
Cough and sputum production
In asthma, cough may be dry or associated with the production of clear or discoloured sputum. Dry cough commonly accompanies shortness of breath, chest tightness and wheeze, and is thought to be related to airway narrowing although the exact mechanisms in relation to airway remodelling remain unclear. The secretion of mucus into the airway lumen will also cause symptoms based on the resultant narrowing of the airway lumen, as will be discussed in the following section. Stimulation of cough receptors in the central airways may be important. However, it is also conceivable that stimulation is due to air-trapping/hyperinflation with activation of stretch fibres, some of which may be due to mucus occlusion of the small airways 90. Productive cough in asthma, with phlegm that is normally clear or white in colour, is readily accounted for by the increased volume of goblet cells and submucosal mucous glands 13, 17, 20, 91. Discolouration of the sputum may be due to the accumulation of neutrophils in relation to superimposed infection or, occasionally in noninfectious exacerbations, due to neutrophils or eosinophils. Persistent productive cough is prominent in some patients with asthma and may overlap with syndromes associated with bronchiectasis such as allergic bronchopulmonary fungal disease. As mentioned previously, most asthmatic patients do not spontaneously produce sputum, although the inflammatory cell profiles of spontaneously produced and induced sputum are similar 92.
Daily cough and sputum production, or chronic bronchitis, is a cardinal feature of COPD 5. This symptom was one of the first to be clearly related to airway remodelling. The Reid index, a measure of increased volume of submucosal mucous glands, was shown to correlate with chronic productive cough in a number of studies of COPD and smoking 49, 91, 93–96. However, mucous gland size is not the only determinant of sputum production: Mullen et al. 97 showed that symptoms in smokers with chronic bronchitis were more strongly related to central airway inflammation rather than the Reid index, and Saetta et al. 98 also showed a relationship of chronic bronchitis symptoms to airway inflammation, specifically neutrophils, macrophages and CD8+ lymphocytes.
Wheeze, cough, chest tightness and shortness of breath: mechanisms of airway narrowing
The lumen of an airway can be narrowed due to: 1) accumulation of fluid, cells or mucus; 2) encroachment of a thickened airway wall with or without smooth muscle shortening; 3) shortening of the smooth muscle surrounding the airway; or 4) collapse of the airway wall (fig. 3a–d⇓, respectively).

Schematic cross-section of the airway. Narrowing of the airway lumen may be due to: a) mucus, cells or other material within the lumen, b) thickening of the airway wall that encroaches on the lumen, c) shortening of smooth muscle around the lumen, d) collapse of the airway wall into the lumen.
Accumulation of mucus and cells in the airway lumen
The accumulation of mucus in the airways in cases of asthma is well recognised 11, 13, 19, 99. It is associated with hypertrophy and hyperplasia of epithelial goblet cells (fig. 1⇑) 13, 20 and hypertrophy of submucosal glands (fig. 4⇓) 13, 17. Remodelling of the mucus-secreting glands in asthma will result in increased mucus production, but it is likely that accumulation and pooling of secreted mucus over time is required to produce excessive airway narrowing 19, 100. Smooth muscle shortening around the airway will amplify these effects 101. Although quantitative studies of the airway epithelium in asthma do not show significant differences in epithelial loss in asthma 17, 18, the increased epithelial cells observed in intraluminal mucus 23, in induced sputum, and in bronchoalveolar lavage from cases of mild asthma suggest increased epithelial turnover in asthma. Impaired clearance of mucus is present during exacerbations of asthma 102 and is a particular feature of fatal asthma 19. The rheological properties of mucus are altered in asthma 103, 104 which may lead to reduced clearance and accumulation of mucus within the airways. Although increased luminal mucus is seen in post mortem cases of mild asthma 105 and is almost invariably a feature of cases of fatal asthma 99, increased mucus production is not a common clinical feature of mild-to-moderate asthma. In fact, the collection of spontaneously produced sputum samples from asthma is difficult and induction with agents such as hypertonic saline is often required 106.

Detail of submucosa of a large airway from a case of fatal asthma. Submucosal mucous gland with numerous acini and interspersed intact mast cells (arrows). Tryptase antibody with haematoxylin counterstain. Scale bar = 50 μm.
In COPD, mucus can accumulate in the airway causing narrowing of the lumen 107 and this accumulation has been correlated with the degree of airflow obstruction 27. Cigarette smoking is also associated with changes in the epithelium including goblet cell hyperplasia 29 and metaplasia and inflammation of the submucosal mucous glands 97, 98.
Thickened airway wall encroaching on the airway lumen
Numerous studies have shown that the airway walls are thicker in asthma in both mild and severe cases 32. Thickening of the airway wall in asthma is related to severity and involves all components of the airway, including the epithelium, reticular basement membrane, submucosa, smooth muscle and mucous glands 17. Contributing factors include oedema and bronchial vessel congestion and proliferation 74, 75, 108. Despite these changes, the increased thickness of the airway wall is not usually associated with narrowing of the airway lumen in asthma 109, 110. It is calculated that, by itself, the increased thickness of the airway wall observed in asthma will increase airway resistance by <10% of values observed in nonasthmatic airways 79, 101. Not surprisingly, the majority of individuals with asthma, who have mild-to-moderate symptoms, have lung function that is within the normal range. It seems unlikely that there is a mechanism for sudden thickening or enlargement of the submucosa which might encroach upon the lumen. In particular, unlike in the nose, there does not appear to be any large capacitance vessels in the submucosa in humans with or without asthma 75. There are more 74 and bigger 75 vessels below the airway epithelium in asthma but these have a relatively small space-occupying effect.
In COPD, the lumen of the airways are significantly narrowed, compared with subjects without airflow obstruction and compared with asthma cases 109. Many studies have shown a correlation between abnormal lung function and lumen dimensions of the small airways in patients with COPD 27, 43, 44, 46, 111–114. In general, these studies have shown a reduction in the diameter of small airways or an increase in the number of small airways with reduced internal diameters, compared with controls. These studies confirm the important role of small airways remodelling in COPD 44, 115. Due to the large total cross-sectional area of the small airways 116, considerable inflammation and/or remodelling may occur before symptoms and abnormalities of lung function become apparent 117. There have been few studies of the larger airways in COPD. Tiddens et al. 48 studied cartilaginous airways from patients with airflow obstruction and showed that the thickness of the airway wall, although not the amount of smooth muscle, correlated inversely with measures of airflow. They also showed that these structural and functional changes were related to airway inflammation.
The increased thickness of the airway wall in COPD is not as prominent as that seen in asthma 44. However, in a similar manner to asthma, the increased thickness of the airways in COPD involves the epithelium, reticular basement membrane, ASM and submucosal mucous glands in an inflammatory and fibrotic process 43, 70. The fixed encroachment of the airway wall on the airway lumen in COPD is not reversed by bronchodilators and accounts for at least some of the persistently increased resistance to airflow. Therefore, in COPD, airway remodelling increases the thickness of the airway wall and directly results in airway narrowing. In contrast, in most (but not all) cases of asthma the increased thickness of the airway wall is greater than that in COPD but the effect on luminal size is much less, so that the functional effects of airway remodelling are only manifest with smooth muscle shortening. Of course increased airway wall thickness may also result in fixed airway narrowing and irreversible airflow obstruction in some patients with asthma 118.
Smooth muscle shortening
Shortening of the smooth muscle around the airway causes narrowing of the lumen. The amount of narrowing that occurs is dependent on three factors: 1) the proportion, relative to the resting length, that the muscle shortens; 2) the proportion of the airway perimeter that is made up of smooth muscle; and 3) the thickness of the airway wall (including smooth muscle, submucosa, epithelium and surface mucus and fluid) within the outer boundary of the smooth muscle layer 101. The relationship of airway lumen size to smooth muscle shortening and wall thickness has been explored by Moreno et al. 101 and expanded in a number of mathematical models 119–121 and is discussed in more detail elsewhere in this series. In asthma, the increased thickness of the airway wall has a space-occupying effect that encroaches on the lumen, minimally (asymptomatic) at rest and markedly (mild to severe symptoms) when the surrounding smooth muscle shortens, even if the degree of shortening is the same as that in nonasthmatic airways 79, 101, 119, 122. It should be pointed out, however, that there may be opposing effects, due to the increased load associated with increased airway wall thickness, on the relationship between changes in lung function and dose of inhaled stimulus, as suggested by the study of Niimi et al. 123.
The loads that oppose shortening of ASM may be critical in determining how much the airway wall can encroach on the airway lumen as the smooth muscle develops force 101. These include external (to the smooth muscle layer) and internal (within the smooth muscle layer) loads. External loads include the mechanical properties of the airway wall or surrounding lung parenchyma that resist deformation. The structural properties of the airway wall and how these may change in asthma are dealt with in the subsequent section.
Loss of elastic recoil of the lung parenchyma has been observed in the lungs of some patients with asthma, even after successful treatment has restored normal lung function 124–126. The structural bases for these changes are not known, although loss of alveolar attachments and peribronchiolar damage to elastin has been reported in cases of fatal asthma 64. Abnormal elastic recoil of the lung, present in many subjects with moderate or severe persistent asthma, has not been associated with abnormalities of the lung parenchyma assessed by computed tomography (CT) scan or measurement of diffusing capacity 127. Although elastic recoil of the lung in patients with asthma has not been shown to be related to duration or severity of asthma 128, abnormal elastic recoil has been identified as a risk factor for near fatal asthma 129.
Less is known about the internal loads that limit muscle shortening, but it is likely that remodelling has the potential to greatly alter them. The internal mechanical properties of structures within the smooth muscle 130–132 may determine both maximum shortening and the velocity of shortening, both of which will influence maximal airway narrowing. In addition, the mechanical properties of increased ECM surrounding smooth muscle cells 62 will affect smooth muscle shortening by altering the freedom of movement between cells and matrix and by constraining muscle cell width 133.
Airway wall collapse
The compliance of the airway wall will influence the loads that oppose ASM shortening. The mechanical properties of the airway wall are partly determined by the properties of the constituents including the epithelium, ECM, glands, blood vessels and cartilage (where present), by the tone of the ASM 134 and possibly by the dynamic changes in fluid and matrix interactions, as seen in articular cartilage 62. The study of Brackel et al. 135 showed that the compliance of the central airways (from the trachea to the mid-part of the right lower lobe) was reduced in patients with asthma. Reduced distensibility of the airways has been observed in patients with asthma 136, has been related to the thickness of subepthelial fibrosis 137 and has been attributed to increased numbers of fibroblasts 54 and collagen deposition 52, 54, 138, 139 below the basement membrane in asthma. The increase in submucosal collagen in asthma is more likely to resist airway dilatation than compression, due to the fibrillar nature of collagen 140.
It is also possible that changes in the ECM within the airway wall and in the external loads may increase collapsibility. Disruption of collagen due to the presence of metalloproteinases 141 may alter its mechanical properties. This is supported by in vitro studies showing increased responsiveness of human ASM after treatment with collagenase 131. Loss of elastin may contribute to altered airway mechanics in asthma, possibly increasing deformability, with a tendency towards increased airway narrowing 63, 65. Thus, in some asthma cases the airways could be more collapsible, which will increase the tendency for airway closure.
In addition to persistent narrowing of the airways in COPD, there is a component of dynamic collapse that further increases airway narrowing and limits maximal airflow 142. This is probably due to loss of elastic recoil of the lung parenchyma 125 and loss of attachments to the airway wall 143 but may also be due to reduced stiffness of the airway wall. Tiddens et al. 134 studied isolated airways from smokers who underwent surgery. They found that the amount and tone of the smooth muscle, but not total airway wall area, were determinants of airway mechanical properties including hysteresis, compliance and collapsibility. In an in vitro study, Opazo Saez et al. 144 found that small airways from smokers with airflow obstruction had more smooth muscle and were able to generate more force and stress (force/cross-sectional area of ASM) in vitro in response to acetylcholine than those from smokers without airflow obstruction. Therefore, it is likely that in COPD, if airway wall compliance is normal or decreased, that smooth muscle stimulation will lead to increased airway narrowing. These studies show that interactions between the ECM and smooth muscle within the airway wall in subjects with COPD will determine the mechanical properties of the airway wall and the degree to which the airway lumen can narrow, either passively or as a result of smooth muscle shortening.
Loss of parenchymal support is evident in patients with COPD where lung recoil is reduced due to emphysema 145, 146 and where the number of alveolar attachments around the airways is reduced due to emphysematous destruction and peribronchiolar inflammation 143. Loss of alveolar attachments has been related to decreased airflow 147, to the reduced bronchodilating effect of a deep inspiration 148, 149 and to abnormal distribution of ventilation 150.
AIRWAY HYPERRESPONSIVENESS
Early dissertations on asthma described the characteristic increased sensitivity of asthmatic patients to inhaled allergens and other irritants 151 that has become known as AHR and has been shown to be a diagnostic feature of asthma 152–154. An early study showed that AHR was related to severity of symptoms over long periods (months) rather than those in more recent days or weeks 155, suggesting that AHR is at least partly determined by remodelling, which probably only changes over long periods 156–158. It should be recognised however, that some factors which influence AHR may change acutely with seasonal asthma 159 and following viral infections 160. Similarly, the reversibility of airflow obstruction by bronchodilators or in response to corticosteroid therapy has been used as a definition of asthma 153, differentiating it from COPD. AHR in asthma can be demonstrated to a wide range of stimuli such as methacholine, histamine, cold/dry air, exercise, nonisotonic stimuli or specific allergens to which the patient is sensitised 161. The degree of AHR is loosely related to clinical severity of asthma, although not to baseline lung function, and severe AHR may be seen in patients with normal lung function 153.
The dose–response curve: its relationship to remodelling
It has been suggested that different aspects of airway wall remodelling may independently influence the sensitivity (position relative to the horizontal/dose axis), the reactivity (slope of the linear part of the curve) and the maximum response 162 of the dose–response curve. For example, inflammation and altered permeability of the airway wall may influence the sensitivity, whereas thickness of the airway wall may influence the reactivity and maximal response. Increasing the deposition of an inhaled stimulus may increase the sensitivity of the response without changing the maximal response 163. Niimi et al. 123 assessed airway inflammation using induced sputum, and airway wall thickness using CT scans in subjects with asthma and related these measures to aspects of the dose–response to inhaled methacholine. While sputum eosinophils were related to airway sensitivity, it was found that reactivity was inversely related to airway wall thickness. The reduced reactivity of the response may have resulted from increased stiffness of the thicker airway wall. An alternative explanation is that reduced access to the smooth muscle by the inhaled methacholine due to increased wall thickness may have reduced the stimulation of the smooth muscle. The findings of Niimi et al. 123 highlight the need to consider not only the space-occupying effects of remodelling, but also the changes in the mechanical properties that remodelling may induce. Little et al. 164 found no correlation between AHR sensitivity and increased airway wall thickness assessed by CT scan. Boulet et al. 165 found a correlation between airway wall thickness measured on CT scan and methacholine sensitivity in patients with asthma who had irreversible airflow obstruction, but not in patients with asthma who had near-normal lung function. Therefore, the reported data on the relation of airway structure by CT scan and AHR remain somewhat conflicting.
AHR and remodelling in asthma
Some studies suggest that easier access of the stimulus to epithelial and submucosal sites may enhance AHR. Ohashi et al. 166 found a positive correlation between the loss of epithelial tight junctions and AHR, while the number of epithelial cells in bronchoalveolar lavage fluid has also been reported to correlate with AHR 20. Jeffrey et al. 15 observed an association of epithelial loss and AHR although this was not replicated in another study 16. AHR has also been shown to be related to the thickness of the reticular basement membrane measured on biopsy 26, 158, 167–169. However, a number of studies have not shown such a relationship 139, 170. The number of subepithelial fibroblasts was suggested to be related to AHR in one study 171. The results of studies correlating ASM function measured in vitro with airway responsiveness measured in vivo have varied 172–176. Few studies have attempted to correlate smooth muscle dimensions with AHR. Woodruff et al. 35 studied smooth muscle in biopsies from asthmatic and nonasthmatic subjects. Although they found that the volume fraction of smooth muscle in the submucosa did not correlate with AHR when the asthmatic subjects were analysed separately, there was a significant correlation when control and asthmatic subjects were analysed as a group. This supports the suggestion that the amount of smooth muscle has a direct effect on AHR in asthma. The results of many studies have not shown consistent relationships between airway remodelling and AHR. It is possible that this variation may be due to spurious relationships between individual elements of remodelled airways and the integrated behaviour of the remodelled airways that results in AHR.
COPD, AHR and remodelling
AHR to inhaled stimuli such as histamine and methacholine occurs in patients with COPD 177. In contrast to patients with asthma without fixed airflow obstruction, the response to bronchoconstrictors is related to the degree of baseline airflow obstruction in COPD 9, 178, 179. This is predictable since the response is measured as a percentage change from baseline and, by definition, patients with COPD start with a lower baseline level of lung function. Therefore, it is likely that the AHR seen in COPD simply reflects normal smooth muscle shortening around an airway that is already narrowed due to airway wall remodelling. This is supported by the study of Riess et al. 180 who showed that airway responsiveness in 77 smokers who underwent lung resection was negatively related not only to baseline lung function, but also to lung recoil, and was positively related to airway wall thickness. These findings support theoretical models based on measured airway dimensions 140. The bronchodilator response has also been examined in COPD and was negatively related to bronchiolar cell metaplasia and emphysema, although not related to ASM 181. In a study that compared patients with centrilobular emphysema and patients with panlobular emphysema, Finklestein et al. 169 found that only those with centrilobular emphysema showed a relationship between wall thickness and airway responsiveness. Therefore, as in asthma, increased airway wall thickness could contribute to AHR in COPD by exaggerating the effects of smooth muscle shortening. However, in COPD, airway remodelling also contributes to reduced baseline airway calibre which in itself artefactually influences AHR. Finally, reduced lung recoil may also contribute to AHR in COPD.
AHR: asthma versus COPD
There are a number of differences in AHR between asthma and COPD. The dose–response curves are generally more left-shifted (increased sensitivity) in patients with asthma, compared with patients with COPD 182. AHR in patients with asthma is characterised by the loss of the maximal or plateau response that is observed in normal subjects 182, 183. One study has shown that plateau responses occur in patients with COPD 184 and another has not 185. In addition, while patients with asthma respond to both direct smooth muscle/nerve stimulation and to indirect stimuli, patients with COPD generally respond only to direct smooth muscle stimulation with histamine or methacholine 9, 184. The differences between AHR in asthma and COPD are intriguing, because if stimulated airway segments from patients with COPD can produce more force than in nonobstructed patients, as discussed previously 144, then all subjects with COPD should have a loss of plateau response or demonstrate AHR to direct and indirect smooth muscle stimuli in vivo. The differences may lie in the relative mechanical properties of the smooth muscle (including length-tension and velocity of shortening considerations) and the airway wall in asthma and COPD. Other factors unrelated to remodelling, such as specific sensitisation, allergic airway inflammation, airway permeability and local neuro-humoral responses also need to be considered. Differences in the responses to adenosine between smokers and nonsmokers with COPD 186 suggest that inflammation may also modulate airway responses in some COPD patients.
CLINICAL SEVERITY OF AIRWAY DISEASE
Although many factors contribute to asthma severity, remodelling is almost certainly important. CT scans and morphometric studies of the proximal airways in asthma have shown a relationship between increased severity of disease and increased thickness of the airway wall 17, 110, 164, 187. Pathological changes include thickness of the total airway wall, inner airway wall, the smooth muscle and mucous glands 17, 188. Deposition of ECM proteins such as collagen 188 results in increased thickness of the reticular basement membrane in asthma, which has been shown to be related to severity in most 170, 189, 190 but not all 139 studies. More severe cases have been characterised by increased ASM 17, 188. The degree of air-trapping, or hyperinflation (measured physiologically or radiologically), and unspecified remodelling also appear to contribute to asthma severity 170. Finally, it is likely that genetic influences help determine the severity of asthma specifically through effects on airway remodelling 191, 192 and/or smooth muscle function 193.
There is a reproducible relationship between the clinical severity of asthma and AHR 152, 194. To the extent that airway remodelling contributes to AHR as discussed previously, more marked changes of remodelling will result in more severe asthma. However, this is only a broad generalisation, since some aspects of remodelling may exaggerate airway narrowing whereas others may protect against airway narrowing (see below).
FIXED AIRFLOW OBSTRUCTION AND DECLINE IN LUNG FUNCTION IN ASTHMA
Fixed airflow obstruction
Asthma is associated with reduced lung function in large population studies, even those where most subjects have mild asthma 195–198. The effect is more marked in asthmatics with more severe disease (fig. 5⇓) 197 and in those with persistent symptoms 195. Remodelling may result in abnormal lung function in patients with asthma early in life (or at the onset of disease) by preventing full lung growth or by accelerating the decline in lung function with age (see below). It should be noted that some patients with asthma also develop fixed airflow obstruction with predictable exertional dyspnoea, as is the case in patients with COPD 199, 200. This overlap led Dutch investigators to suggest a common link between asthma and COPD and the hypothesis that smokers with features of asthma would be more likely to develop COPD 201.

Data from the Melbourne cohort study 197 showing the per cent predicted (% pred) value of the forced expiratory volume in one second (FEV1) from childhood to middle-age for subjects classified by status at first study as nonasthmatic (□), mild wheezy bronchitis (•), wheezy bronchitis (▴), mild asthma (♦) and severe asthma (▪). The differences in lung function between the groups remained much the same over the course of 35 yrs.
The reduction in lung function in asthma appears to be related to duration and clinical severity 199, 200, 202–205. In some studies of chronic, severe asthma, persistent airflow obstruction has been shown to be related both to inflammation, based upon exhaled nitric oxide and blood eosinophilia, and to remodelling, shown as increased wall thickness on CT scan 206. Similarly, increased wall thickness on CT scan has been related to a decrease FEV1 in asthma in some studies 110 but not others 164.
Decline in lung function
The rate of decline in lung function in adult patients with asthma appears to be increased relative to those without asthma 198, 207–209. The Busselton health studies, which have followed a general population cohort since 1966, showed an increased rate of decline in FEV1 in asthmatic subjects 208, as was seen in the Copenhagen health studies 207, which also demonstrated that the decline in asthma was independent of cigarette smoking. Further study of the Busselton population 198 agreed with these findings but also showed that lung function at age 20 yrs was reduced in asthmatics but not in smokers (fig. 6⇓). Some individuals with asthma may be genetically predisposed to a more rapid decline in lung function 210, 211 although the pathogenetic pathways related to this assocaition are not established.

Data from the Busselton Health Study in a) males and b) females, showing height-corrected forced expiratory volume in one second (FEV1) versus age for nonasthmatic nonsmokers (–––), asthmatic nonsmokers (······), nonasthmatic smokers (– – –) and asthmatic smokers (- - -). Subjects who have asthma and smoke have the fastest rate of decline of FEV1 with age. Smoking is associated with a faster rate of decline compared with nonsmokers, but not a deficit in lung function at age 19. Asthma is associated with both an increased rate of decline of FEV1 and a deficit in lung function at age 19 yrs. Reproduced from 198 with permission.
Risk factors for progressive loss of lung function in patients with asthma include smoking, atopy, adult onset, increased reversibility of obstruction, increased numbers of airway eosinophils and AHR 212. ten Brinke et al. 213 showed that sputum eosinophilia, adult onset of asthma and AHR predicted fixed airflow obstruction in patients with asthma, although only sputum eosinophilia was significant by multivariate analyses. Vonk et al. 214 found a relationship with pre-bronchodilator FEV1, AHR and change in FEV1 for irreversible airflow obstruction. The study of Brown et al. 199 showed that fixed airflow obstruction, present after 2 weeks’ treatment with oral corticosteroids, was related to the duration and severity of asthma. Whether these abnormalities are truly irreversible is questioned by studies that show improvements in lung function with high-dose corticosteroids 215, and highlights the need to distinguish “severe”, “steroid-resistant”, “poorly controlled” and “difficult-to-control” classifications of asthma 216. These considerations are important since Strachan et al. 217 found that lung function (pre- and post-bronchodilator) in a cohort followed from childhood to 35 yrs of age was reduced most in those with persistent wheeze, less in those with intermittent (but current) wheeze and not at all in those with transient wheeze (no reported wheeze in the previous 12 months), which suggests that current symptoms partly account for deficits in lung function and, therefore, apparent increased decline in lung function. A similar conclusion was reached by Ulrik and Lange 209, who found that the greatest decline in lung function occurred in patients with new (recent symptoms) asthma. In this regard, decrements in lung function may result from exacerbations, which may be amenable to treatment 218.
The beginnings of remodelling: longitudinal studies of lung function
The deficits in lung function in subjects with asthma arise early in the course of the disease 196, 197. Rasmussen et al. 196 found that abnormal lung function at age 26 in asthmatics was related to male sex, AHR at age 9 yrs, early onset of asthma and reduced lung function by age 9 yrs. Sears et al. 195, reporting on the same cohort, showed that the risk of persistent wheeze from age 9–26 yrs was related to reduced lung function. In studies of children 196, 197 the group data do not suggest a faster decline in lung function, which supports the argument that deficits in lung function occur early in the disease and then remain stable (fig. 5⇑). However, Covar et al. 210, in a re-analysis of the Childhood Asthma Management Program data, suggested that ∼25% of children had a persistent decline of lung function over the 5 yrs of the study that was not impacted by corticosteroid therapy. These children were younger, more likely to be male and were less atopic. The Tucson study 219 showed that abnormalities of lung function in infancy were more common in children with transient wheeze associated with maternal smoking and viral illness, than in those who later developed asthma. However, infants with persistent wheeze and subsequent asthma initially had normal lung function but developed abnormalities of airway function in association with asthma 219. In a study from Perth, Western Australia, children with abnormalities of lung function and persistent wheeze at age 6 yrs had normal measures of airflow but increased airway responsiveness as infants 220. Therefore, it seems likely that children with asthma may have relatively normal lung function as infants but develop fixed abnormalities of lung function early in the course of the disease, and that some have a progressive decline in lung function.
The beginnings of remodelling: pathological studies
Reticular basement membrane thickness is increased in the airways of children with mild-to-severe asthma, aged 6–16 yrs 221. Biopsies of airways of younger children with wheezing illnesses and abnormal lung function have not shown changes of remodelling although these infants did not clearly have asthma 222. Another study showed that in infants who subsequently developed asthma, airway pathology was evident before the onset of their disease 223, which supports the concept that changes of inflammation and remodelling occur early in the disease, even before symptoms are evident. From the few data available it would appear that remodelling (suggested by increased thickness of the reticular basement membrane) may begin early in asthma that arises in children, and may even precede the onset of symptoms. There are no data regarding the smooth muscle in infants or young children who are destined to develop asthma.
Studies of airway morphology in adult-onset asthma are few with virtually no longitudinal data. In patients with occupational asthma 224 and in elite skiers who develop asthma-like symptoms 225 elements of remodelling are observed, but the relation to onset and duration of disease is less clear. Laprise and Boulet 226 followed up a group of subjects with asymptomatic AHR, and found that those who became symptomatic at follow-up had focal, rather than continuous, thickening of the reticular basement membrane. The reticular basement membrane increased in thickness with the development of symptoms and this was associated with increased numbers of activated CD4+ T cells. In a study of asthmatics who were asymptomatic for some years and not receiving treatment, van den Toorn et al. 227 found increased eosinophil numbers and thickening of the reticular basement membrane, to a level between that of nonasthmatic control subjects and symptomatic cases of asthma.
There are few studies of the effects of age versus duration on airway remodelling. Bai et al. 228 showed that older subjects with asthma had more smooth muscle and a trend towards more ECM, compared with younger subjects with asthma. These studies demonstrate progression of some aspects of airway remodelling in patients with new asthma symptoms and persistence of these changes in the absence of symptoms. The latter suggests that remodelling may remain asymptomatic for some time. The reasons for this are intriguing, although possibilities might include the requirement for an additional factor (such as inflammation), absence of an adequate stimulus to contract smooth muscle or the protective effect of remodelling itself (see below).
FIXED AIRFLOW OBSTRUCTION AND DECLINE IN LUNG FUNCTION IN COPD
COPD is defined by abnormal lung function. The GOLD classification 5, 7 categorises patients on the basis of symptoms and lung function. Previously, stage 0 was characterised by normal lung function but with chronic symptoms of cough and sputum production. Stages 1–4 are characterised as “mild”, “moderate”, “severe” and “very severe”, based upon decreasing levels of lung function. The fixed abnormalities of lung function in COPD are due to a variety of physiological abnormalities that include changes intrinsic to the airway wall as outlined previously. The thickness of the airway wall, including the epithelium, lamina propria, smooth muscle and adventitia are all related to the severity of airflow obstruction in patients with COPD 27, 43, 44, 46, 111–114.
Accelerated loss of lung function develops over time in susceptible smokers 229, 230, and is most likely due to genetic mechanisms 231. Smoking cessation leads to normalisation of the rate of decline of lung function and restarting increases the rate of decline 229, 230. Airway inflammation persists in patients with COPD and chronic bronchitis who have stopped smoking 232. It has been suggested that mucus hypersecretion itself contributes to an increased decline of lung function, even after adjustment for the effects of cigarette smoking 233. Whether this is due to persistent inflammation or progressive remodelling of the airways is not clear. The progressive loss of lung function associated with continued smoking is probably due, in large part, to continued parenchymal destruction by activated neutrophils, compromised lung defence mechanisms such as damage to alveolar type-II cells, and to airway inflammation 234.
AHR has been associated with a faster decline in lung function in subjects with COPD 235–237. This association may be due to progressive airway remodelling, although there are no longitudinal studies in COPD to assess this hypothesis, and other influences such as inflammation must be considered 27. In this regard, the relationship between exacerbations of symptoms and rate of decline in lung function is interesting. It was initially considered that following an exacerbation, subjects with COPD tended to return to the level of lung function that was present prior to the exacerbation 238. More recently, however, there is evidence to show that more frequent exacerbations are associated with a more rapid decline in FEV1 239 and it has been observed that some subjects do not fully recover, even 6 months following an exacerbation 240. The rate of decline may, in part, be due to persistently increased airway inflammation 241 which may be related to reduced viral clearance 242 or to transient increases in inflammation 243, both of which may result in incremental airway remodelling and loss of lung function.
REVERSIBILITY OF REMODELLING AND ABNORMAL LUNG FUNCTION IN ASTHMA AND COPD
The degree to which abnormal lung function and airway remodelling are reversible is unclear. Some studies have shown that prolonged treatment with inhaled corticosteroids may reduce thickness of the reticular basement membrane 157, 158. The use of lower doses and/or shorter periods of treatment has not shown any effect 158, 244. While these changes appear to be related to remodelling processes across the airway wall, including changes in ASM 187, 189, no studies have directly addressed the effect of treatment on ASM volume. Using CT scans, Niimi et al. 245 showed partial reversibility of airway wall thickness in patients with asthma. The authors suggested that there were steroid-responsive (possibly inflammatory) and steroid-unresponsive components (possibly remodelling) that contributed to increased airway wall thickness in asthma. It is generally regarded that remodelling is an irreversible process, but, as suggested previously, this may be related to dose of treatment. Duration of treatment is also likely to be important since improvements in AHR may continue for periods of ≥6 months, despite treatment alleviating symptoms within weeks 156, 158, 246. The study of Ward et al. 158 showed that although inflammation improved after 3 months of therapy, increased thickness of the reticular membrane was improved only at the second time-point of observation, which was 12 months after treatment onset. More studies of this nature are needed to examine the response of airway remodelling to treatment. This will also require the development of less invasive means of monitoring remodelling. Finally, the observations that the resistance of inflammation and remodelling to treatment in patients with COPD may be related to acetylation of DNA 247, shows that there are mechanisms that may account for nonreversible changes that may be targeted in the future.
FATAL ASTHMA
The pathology of asthma was initially described in case series of fatal asthma 13, 248–251. These largely qualitative descriptions highlighted common features, including: the infiltration of the airway wall with eosinophils, neutrophils and lympho-mononuclear cells; occlusion of airway lumens with mucus, cellular debris and eosinophil products (Charcot–Leiden crystals); and thickening of the airway wall with prominence of the smooth muscle and mucous glands both in the epithelium and in the submucosa. The classic publication by Huber and Koessler in 1922 11 was the first to attempt to quantify the degree of remodelling in fatal asthma and showed that the airway wall was thicker, as was the smooth muscle, in both small and large airways in cases of asthma (fig. 7⇓). Airway sizes from cases and controls were matched using the outer diameter of airways cut in transverse section. This might tend to over-estimate the wall thickness in asthma, since it is likely that, in these cases, airways from the same generation may artefactually have smaller outer diameters due to muscle contraction. This problem was overcome later by using the basement membrane perimeter to match airway size 252. However, the results of Huber and Koessler 11 showed large quantitative differences between controls and cases of fatal asthma. The paper (all 92 pages) is still well worth reading!
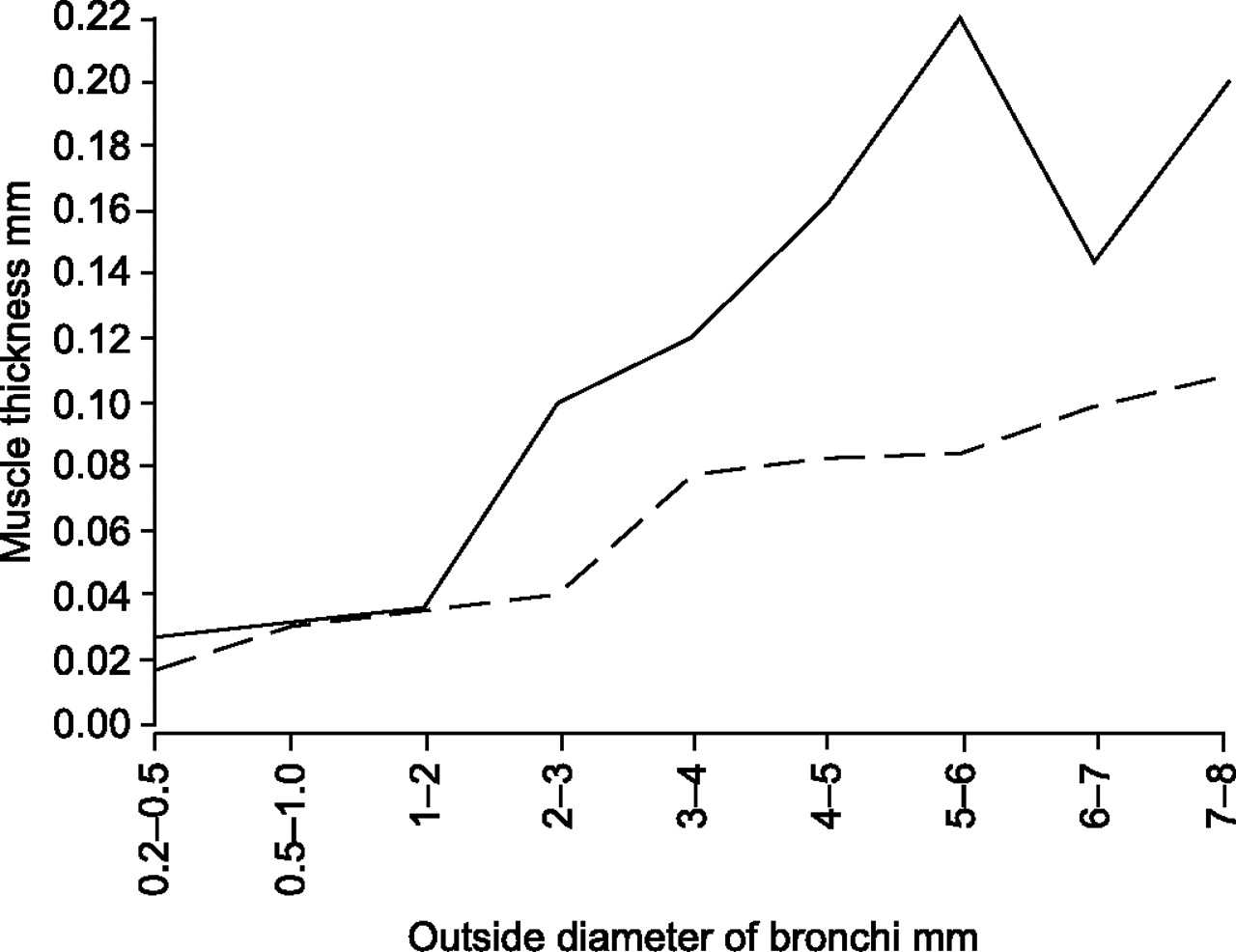
Thickness of airway smooth muscle plotted against outside bronchial diameter, measured in 1922 in post mortem tissues from nonasthmatic subjects (– – –) and in cases of fatal asthma (–––). Modified from 11 with permission.
Since these early studies, quantification of remodelling in fatal asthma 32 has confirmed increased airway wall thickness, increased area of smooth muscle, increased mucus in the airway lumen, increased area of the submucosal mucous glands, increased deposition of ECM proteins and increased number and size of blood vessels within the airway wall, compared not only with nonasthmatic control cases but also with nonfatal, clinically mild-to-moderate cases of asthma. A recent study that used fractal geometry to describe measurements from airway casts showed that fatal asthma is associated with reduced complexity of the bronchial tree compared with control cases 1, although they were not significantly different from nonfatal cases.
Is fatal asthma just severe asthma or is it a distinct entity?
Clinical characteristics of those who die from asthma can be compared with those with near-fatal asthma, those with severe nonfatal asthma and those with mild-to-moderate asthma. The clinical severity of asthma is based upon: symptom frequency during the day or at night; treatment requirements, particularly oral corticosteroids; lung function; the frequency of emergency treatment and attacks (exacerbations); the frequency of admission to hospital; interruption of usual activities by asthma; and the history of near-fatal attacks 253. Comparisons using these parameters suggest, almost universally, that those who die from asthma have a clinical history comparable with cases of severe asthma 17, 99, 254. Comparisons of near-fatal and fatal cases of asthma show similar levels of clinical markers of severity and similar reported psychiatric problems 255, although whether these are higher than the general population (except for depression and anxiety) is controversial 256. Another study showed that sudden onset of symptoms could occur in both groups 257. These observations suggest that overall, clinically severe asthma is not obviously different from fatal asthma. Therefore, additional factors are required for an increased risk of death from asthma 258.
Potential physiological differences between asthma that is severe and asthma that is life-threatening or fatal include the increased response of the forced vital capacity to inhaled bronchoconstrictors in near-fatal asthma, compared with severe but not near-fatal cases, as well as a lower starting level of lung function 259. This finding raises the possibility that remodelling may determine the nature and extent of airway narrowing and, therefore, the degree of the response and the likelihood of a fatal or near-fatal event. Interestingly, one of the strongest risk factors for a near-fatal or fatal asthma attack is a previous near-fatal event, which supports an underlying inflammatory or structural change in the lungs that contributes to the risk. There have been very few studies that have compared the genetics of fatal or near-fatal cases with less severe cases. Although adverse reactions to β-agonists have been suggested to be genetically driven 193, Weir et al. 260 did not find differences between asthma groups of different severity with regard to polymorphisms of the β-adrenergic receptor.
Pathologically, cases of fatal asthma have shown similar changes to those seen in severe asthma. Synek et al. 261 showed increased numbers of eosinophils and epithelial CD3+ cells in large, but not small, airways in fatal asthma compared with mild-to-moderate asthma cases that died from nonrespiratory causes. No differences in numbers of mast cells, monocyte/macrophages or neutrophils or airway dimensions were reported. Sobonya 262 studied six cases of severe allergic asthma with persistent airflow obstruction that had died from nonrespiratory causes. It was found that severe asthma cases had increased thickness of the reticular basement membrane and two cases of asthma had reduced luminal area of small airways associated with fibrosis, compared with nonasthma controls. In contrast to studies of fatal cases, increased thickness of the airway wall or the ASM was not observed, which suggests this factor may differentiate fatal from severe asthma, but further studies comparing airway dimensions in cases of clinically severe (but not fatal) asthma with cases of fatal asthma would better answer this question.
Is fatal asthma an extreme exacerbation?
Exposure to a number of stimuli can result in exacerbations of asthma and, in some cases, death. These include viral respiratory infection 263, 264, allergens such as Alternaria 265, soybeans 266, pollen exposure associated with thunderstorms 267, 268 and specific occupational exposures 269. These exposures will induce airway narrowing, the magnitude of which may be proportional to the underlying airway remodelling. In support of this, Cockcroft and co-workers 270, 271 twice demonstrated that the response to inhaled allergen is related not only to the degree of allergic sensitisation shown by the skin weal response, but also to the degree of remodelling shown by the nonspecific response of the airways to inhaled histamine. The effects of stimulus strength also agree with responses predicted by models using airway dimensions of asthmatic and nonasthmatic subjects 101, 272.
What features of remodelling contribute to a fatal attack?
A number of studies have examined cases of fatal asthma in relation to the duration of the attack 273–276. All studies showed a clear dichotomy of times to death from the onset of symptoms of the fatal attack and consistent differences in the ratio of numbers of eosinophils to neutrophils in the airway walls. Short-course cases demonstrated a decreased ratio of eosinophils to neutrophils, compared with long-course cases. This led Sur et al. 274 to postulate that a specific phenotype of asthmatic exacerbation that responded with an influx of neutrophils was predisposed to sudden death from asthma. Carroll et al. 275 observed that the mucous gland area was greater in the short-course cases, and proposed that increased airway remodelling contributed to a rapid death after exposure to an inflammatory stimulus and that the decreased eosinophil-to-neutrophil ratio was due to capturing the early neutrophilic response that is characteristic of the usual response to an inflammatory stimulus 277. It should be noted that the ratios of neutrophils to eosinophils will also be partly determined by the day-to-day level of eosinophilic inflammation present in the chronic state.
Multiple additional pathological features have been observed in fatal asthma. Luminal mucus and muscle shortening are likely to contribute to asthma death, as noted in studies where the lungs were not fixed in inflation 99, 258. Increased shortening of ASM persisted despite increased post mortem serum levels of salbutamol in the short-course cases and there was more mucus accumulation in the airway lumens of long course-cases 276. Finally, Mauad et al. 64 reported loss of alveolar attachments with accompanying loss of small airway elastin in fatal asthma cases, perhaps leading to airway closure due to loss of parenchymal tethering. Thus, different mechanisms of excessive airway narrowing (mucus accumulation, loss of alveolar attachments or smooth muscle shortening) may account for death in fatal asthma.
IS REMODELLING ONLY DELETERIOUS OR CAN IT BE PROTECTIVE?
This question was recently addressed in a review by McParland et al. 278. Much of the previous discussion suggests that remodelling may only have deleterious effects, such as excessive airway narrowing and symptoms, increased airway responsiveness, fixed airflow obstruction and accelerated decline in lung function. However, it should be considered that the increased thickness of the airway wall, smooth muscle, mucous glands and deposition of ECM proteins may have beneficial effects, without which the effects of airway inflammation or the response of the airways to bronchoconstricting stimuli such as allergen would be even more detrimental to the patient. It is conceivable that an initial abnormality that impacts on airway structure and/or function is followed by a secondary remodelling process which minimises the effect of the initial change 279. The latter may occur over a short or long period but may, in fact, be protective (fig. 8⇓). Primary events might include increased ASM that is present at birth or develops within the first few years of life or to airway inflammation due to hypersensitivity to inhaled stimuli or an inherent abnormality of the epithelium 280.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram of the relationships between remodelling that may result in symptoms and remodelling that may prevent or minimise symptoms. ASM: airway smooth muscle; AHR: airway hyperresponsiveness; ECM: extracellular matrix.
These events may give rise to secondary protective events. The increase in ECM that occurs in asthma could serve to minimise shortening by adding to (rather than reducing) the loads against which smooth muscle shortens, thereby limiting airway narrowing. Such loads might include increased stiffness of the airway wall and increased series elastic loads that inhibit mucosal folding 41, 281. McParland et al. 282 showed that loads opposing shortening of ASM might be substantial in relation to the ability of the smooth muscle to generate force. In addition, Milanese et al. 283 showed an inverse relationship between thickness of the reticular basement membrane and airway responsiveness. The increase in ECM around bundles of ASM 62 may result in radial constraint that prevents thickening of smooth muscle cells as they shorten 133. The distensibility of the airways is generally decreased in asthma 135–137 which may indicate increased airway wall stiffness and resistance to deformation. Niimi et al. 123 showed an inverse relationship of airway reactivity (but not sensitivity) to airway wall thickness seen on CT scan. This study supports the concept that airway wall thickening in asthma may serve as a protective response, at least in milder cases.
There are few studies addressing these relationships in COPD. The most relevant is the study of Colebatch et al. 284 which showed that some patients with COPD and “primary narrowing of airways” had reduced airway conductance relative to elastic recoil of the lung, suggesting reduced airway distensibility. Whether this equates to a reduced tendency to collapse was not examined. Tiddens et al. 285 examined in vivo lung function in patients undergoing surgery and found that airway collapsibility, estimated in vivo as the transpulmonary pressure at zero flow, was not related to the level of pre-operative airflow obstruction or to airway wall dimensions, measured post-operatively in airways from the resected specimen of lung. In a similar study, Tiddens et al. 134 found that although the airway wall thickness was related to measures of airflow obstruction it was not related to in vitro measurements of collapsibility of airway segments. This suggests that although remodelling reduces airflow and airway distensibility in COPD, it does not resist collapse of the airway and therefore does not have a protective role.
More studies are needed to try and establish the relative importance of remodelling as protective or deleterious. These studies will require the use of reliable but noninvasive methods of monitoring to allow longitudinal studies of the effects of interventions on remodelling and lung function. These studies are necessary because treatments aimed at remodelling may potentially reverse both harmful and helpful aspects of remodelling.
CONCLUSION
Although airway remodelling is closely related to airway inflammation, it has its own consequences with regard to symptoms, exacerbations (including fatal attacks of asthma), loss of lung function, decline in lung function and response to treatment. However, the specific elements of remodelling which contribute to the symptoms or progression of asthma and COPD remain poorly understood, partly due to the heterogeneity of these diseases and the complex interplay between numerous structural components within the airways. On the one hand, remodelling in asthma is associated with thicker and probably stiffer airway walls which encroach minimally on the airway lumen. The airways resist distension (except in mild cases), but smooth muscle shortening causes exaggerated airway narrowing. On the other hand, airway remodelling in COPD is less marked but encroaches on the airway lumen, limiting airflow and restricting dilatation, but probably not collapse. Changes in the epithelium, the smooth muscle and various ECM proteins appear to contribute to airway remodelling in both asthma and COPD. The relative amounts and interactions of these tissues are likely to account for the different natural histories of these two conditions.
Future directions for research in this area include: 1) establishing methods for assessing remodelling so that it can be more readily related to clinical aspects of airway disease, lung function, airway inflammation and responsiveness to treatment; 2) identifying the specific elements that contribute to airway remodelling, with particular with regard to the extracellular matrix (this seems to be an expanding area of current research); 3) comparing elements of remodelling in patients with chronic obstructive pulmonary disease with those in patients with asthma (who may or may not smoke cigarettes) who have fixed airflow obstruction; and 4) assessing longitudinal changes in remodelling.
Acknowledgments
The authors wish to thank P. Maxwell (West Australia Sleep Disorders Research Institute, Perth, Australia) for reading drafts of the manuscript and preparation of the figures, N. Carroll (State Department of Sport and Recreation, Mandurah, Australia) for helpful comments on earlier drafts of the manuscript and C. Robertson (Dept of Respiratory Medicine, Royal Children’s Hospital, Parkville, Victoria, Australia) who provided figure 5.
Footnotes
-
Previous articles in this series: No. 1: Fixman ED, Stewart A, Martin JG. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J 2007; 29: 379–389. No. 2: Bergeron C, Tulic MK, Hamid Q. Tools used to measure airway remodelling in research. Eur Respir J 2007; 29: 596–604. No. 3: Lloyd CM, Robinson DS. Allergen-induced airway remodelling. Eur Respir J 2007; 29: 1020–1032.
- Received December 13, 2005.
- Accepted October 2, 2006.
- © ERS Journals Ltd
References
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











Jump To
- Article
- Abstract
- REMODELLING: THE CHANGES IN ASTHMA AND COPD
- EVIDENCE THAT REMODELLING INFLUENCES THE CLINICAL EXPRESSION OF AIRWAY DISEASES
- AIRWAY HYPERRESPONSIVENESS
- CLINICAL SEVERITY OF AIRWAY DISEASE
- FIXED AIRFLOW OBSTRUCTION AND DECLINE IN LUNG FUNCTION IN ASTHMA
- FIXED AIRFLOW OBSTRUCTION AND DECLINE IN LUNG FUNCTION IN COPD
- REVERSIBILITY OF REMODELLING AND ABNORMAL LUNG FUNCTION IN ASTHMA AND COPD
- FATAL ASTHMA
- IS REMODELLING ONLY DELETERIOUS OR CAN IT BE PROTECTIVE?
- CONCLUSION
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics