





Abstract
BIBF 1000 is a small molecule inhibitor targeting the receptor kinases of platelet-derived growth factor (PDGF), basic fibroblast growth factor and vascular endothelial growth factor, which have known roles in the pathogenesis of pulmonary fibrosis.
The anti-fibrotic potential of BIBF 1000 was determined in a rat model of bleomycin-induced lung fibrosis and in an ex vivo fibroblast differentiation assay. Rats exposed to a single intra-tracheal injection of bleomycin were treated with BIBF 1000 starting 10 days after bleomycin administration. To gauge for anti-fibrotic activity, collagen deposition and pro-fibrotic growth factor gene expression was analysed in isolated lungs. Furthermore, the activity of BIBF 1000 was compared with imatinib mesylate (combined PDGF receptor, c-kit and c-abl kinase inhibitor) and SB-431542 (transforming growth factor (TGF)-β receptor I kinase inhibitor) in an ex vivo TGF-β-driven fibroblast to myofibroblast differentiation assay, performed in primary human bronchial fibroblasts.
Treatment of rats with BIBF 1000 resulted in the attenuation of fibrosis as assessed by the reduction of collagen deposition and the inhibition of pro-fibrotic gene expression. In the cellular assay both SB-431542 and BIBF 1000 showed dose-dependent inhibition of TGF-β-induced differentiation, whereas imatinib mesylate was inactive.
BIBF 1000, or related small molecules with a similar kinase inhibition profile, may represent a novel therapeutic approach for the treatment of idiopathic pulmonary fibrosis.
Fibrotic conditions can occur in all tissues but are especially prevalent in organs that have frequent exposure to chemical and biological insults, e.g. the lung, skin, digestive tract, kidney and liver 1–3. These conditions often compromise the normal function(s) of the organ and many fibrotic diseases are at least severely debilitating, if not life threatening 4.
Fibroses of the lung represent a set of pathological changes which accompany a wide range of inflammatory conditions of the conducting airways. For instance, in patients with chronic obstructive pulmonary disease (COPD) a patchy alveolar wall fibrosis with peribronchiolar distribution is present, whereas in patients with chronic asthma, fibrosis is predominantly localised to the lamina reticularis, resulting in a thickening of the basement membrane 5, 6. In both conditions a continuously ongoing inflammation–repair cycle in the airways leads to permanent structural changes in the airway wall (remodelling), of which interstitial collagen fibrosis is the major component 7, 8. Similar aetiologies have been observed in the liver 9. In contrast to the fibrotic changes observed in COPD and asthma, in patients with diseases such as idiopathic pulmonary fibrosis (IPF) and acute respiratory distress syndrome (ARDS), the fibrotic changes are more severe and widely disseminated. In these diseases, fibrosis is associated with extreme morbidity and the clinical course is invariably one of gradual deterioration. Median length of survival from time of diagnosis ranges 2.5–3.5 yrs 4, 10.
Although the degree of pulmonary fibrosis differs between various lung diseases, there is evidence to suggest that the underlying pathophysiological mechanisms involved in development may be similar across diseases. In all forms of pulmonary fibrosis, fibroblasts and myofibroblasts are the most predominant cells 11, 12. Both cell types become activated by growth factors secreted by the airway epithelium after the inflammatory damage 13, 14. Depending on the precise stimulatory milieu, fibroblasts either transform to myofibroblasts or proliferate, resulting in areas of fibroblastic foci which are thought to be the sites of active extracellular matrix (ECM), collagen and fibronectin synthesis, and are regarded to be the leading edge of fibrosis 15, 16.
The polypeptide mediators and growth factors believed to be pivotal for the fibrotic process include transforming growth factor (TGF)-β, vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF)-2, platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), insulin-like growth factor (IGF), epidermal growth factor, chemokine ligand-18 and endothelin (ET)-1 17–26. Amongst these, TGF-β is believed to be a critical mediator of fibrogenesis by exerting immunological actions, having direct effects on structural cells involved in the synthesis of ECM and affecting fibroblast proliferation and the differentiation of fibroblasts into myofibroblasts 27, 28. Several pre-clinical studies have shown that inhibition of TGF-β-signalling results in attenuation of fibrosis in rodents 29–30, suggesting that drug-targeting of the TGF-β pathway could provide a useful therapeutic intervention in human fibrotic diseases including IPF. Unfortunately, TGF-β is a pleiotropic mediator and a number of reports have suggested that anti-TGF-β therapy may result in a number of unacceptable adverse effects 31, 32, particularly, tumour promotion.
Another important fibrogenic mediator, PDGF, induces fibroblast chemotaxis, fibroblast proliferation and promotes fibroblast-mediated tissue matrix contraction 33. Furthermore, a number of fibrogenic mediators, such as TGF-β, interleukin (IL)-1, tumour necrosis factor-α, bFGF and thrombin, exhibit PDGF-dependent pro-fibrotic activities 34–38. Two isoforms of PDGF, namely PDGF-C and PDGF-D, are increased in expression during bleomycin-induced lung fibrosis and it has been shown that PDGF receptor tyrosine kinase inhibitors markedly attenuate radiation-induced pulmonary fibrosis 34, 39–41.
Fibroblasts isolated from patients with moderate-to-severe asthma have the ability to transform into myofibroblasts after in vitro stimulation with TGF-β resulting in the secretion of VEGF, FGF and ET-1 42. ET-1 is a known potent mitogen for smooth muscle cells and is thought to be responsible for the increased smooth muscle mass in patients with chronic inflammation of the lungs. Both VEGF and bFGF-2 are elevated in patients with asthma and are associated with increased vascularity 43, 44. Transfection of the soluble VEGF receptor (sflt-1) gene resulted in attenuation of pulmonary fibrosis in a mouse model of bleomycin-induced pneumopathy, suggesting that an anti-VEGF approach might also offer a suitable anti-fibrotic therapy 18.
The present authors have recently shown in the bleomycin-induced lung fibrosis model in rats that an initial inflammatory phase is followed by subsequent fibrosis. Depending on the treatment scheme, anti-inflammatory and anti-fibrotic activities of test compounds can be discriminated in this model 41. Using this model, it was shown that a prototype anti-inflammatory treatment (the oral steroid prednisolone) attenuated lung fibrosis when commenced at day one, but had no efficacy if administered from day 10 onwards. In contrast, treatment with a prototype anti-fibrotic compound (oral imatinib mesylate, a c-abl/c-kit/PDGF receptor kinase inhibitor) was effective, even when administered beginning at day 10, post-bleomycin treatment 41.
The present study used BIBF 1000, a prototypical small molecule inhibitor selective for the family of VEGF, FGF and PDGF receptor tyrosine kinases 45 and its activity was studied in the aforementioned therapeutic bleomycin model and in an ex vivo assay of pulmonary fibrosis. BIBF 1000 is shown to attenuate fibrosis in vivo and inhibit the differentiation of fibroblasts to myofibroblasts in vitro, indicating that this class of compounds may be useful for the treatment of IPF while avoiding the possible adverse effects of direct TGF-β inhibition.
MATERIALS AND METHODS
Compounds
Imatinib mesylate (Novartis, Basel, Switzerland) and bleomycin sulfate (HEXAL, Holzkirchen, Germany) were purchased from a local pharmacy. BIBF 1000 45 was synthesised by the department of chemistry, Boehringer Ingelheim (Biberach an der Riss, Germany). SB-431542 46 is available from Sigma-Aldrich (Schnelldorf, Germany). Recombinant TGF-β1 (Serotec, Raleigh, NC, USA) and TGF-β2 (Sigma-Aldrich) were diluted with sterile water and stored in siliconised tubes (Eppendorf, Hamburg, Germany).
Bleomycin administration and treatment protocol
All experiments were performed in accordance with German guidelines for animal welfare and were approved by the responsible authorities.
A dose of 2.2 mg·kg−1 bleomycin sulfate was determined to be efficacious in establishing IPF 41. At day zero, male Wistar rats (10 per group) were intra-tracheally injected with bleomycin sulfate in 300-μL saline using a catheter (0.5 mm internal diameter, 1.0 mm external diameter) through the nasal passage. To determine the fully effective dose of BIBF 1000, rats treated with 2.2 mg·kg−1 bleomycin were administered BIBF 1000 (10, 30 and 50 mg·kg−1 in 1 mL 0.1% Natrosol (Merck KG, Darmstadt, Germany)) from day 0–21 and fibrotic markers were analysed in lungs isolated at day 21. The most efficacious dose was 50 mg·kg−1, showing complete inhibition of bleomycin-induced fibrosis. At none of the applied doses did the animals show any signs of toxicological side-effects.
For the experiments detailed in the present study, BIBF 1000 (50 mg·kg−1) was orally administered once daily from day 10–21, after which the rats were sacrificed and the lungs excised. Control rats were treated on day zero with saline only (saline group) and rats treated with bleomycin received vehicle alone from day 10–21 (bleomycin group). The degree of fibrosis was analysed again by gene expression profiling and histology of the excised lungs.
Histology
Histology was performed as previously described elsewhere 41. Collagen deposition was assessed using Masson's Trichrome staining as previously described 41, 47.
Total RNA extraction and synthesis of cDNA
Total RNA extraction and synthesis of cDNA was carried out using previously published methods 41.
Investigation of gene expression using real-time PCR
Gene expression was investigated using previously published methods 41. Primers for the 18S endogenous control and TGF-β1 were purchased as pre-developed assay reagent kits (Taqman®; Applied Biosystems, Foster City, CA, USA). Primers and probes for pro-collagen I, CTGF and fibronectin were designed using PrimerExpressTM (Applied Biosystems). At least one of the primers or probes in each set overlapped an intron/exon junction, thus eliminating the possibility of amplifying any contaminating genomic DNA in the cDNA sample. The following primer and probe sequences were used. Rat fibronectin forward (F): 5′-GATGCCGATCAGAAGTTTGGA-3′; reverse (R): 5′-TCGTTGGTCGTGCAGATCTC-3′; probe (Pr): 5′-FAM-CTGCCCAATGGCTGCCCATGA-3′TAMRA. Rat pro-collagen F: 5′-CAGACTGGCAACCTGAAGAAGTC-3′; R: 5′-TCGCCCCTGAGCTCGAT-3′; Pr: 5′-FAM-CTGCTCCTCCAGGGCTCCAACGA-3′TAMRA. Rat CTGF F: 5′-CGCCAACCGCAAGATTG-3′; R: 5′-TACACGGACCCACCGAAGAC-3′; Pr: 5′-FAM-CGTGTGCACTGCCAAAGATGGTGC-3′TAMRA. Human CTGF F: 5′-GCGGCTTACCGACTGGAA-3′; R: 5′-GGACCAGGCAGTTGGCTCTA-3′; Pr: 5′-FAM-CACGTTTGGCCCAGACCCAACTATGA-3′TAMRA. Human α-smooth muscle actin (SMA) F: 5′-GACAGCTACGTGGGTGACGAA-3′; R: 5′-TTTTCCATGTCGTCCCAGTTG-3′; Pr: 5′-FAM-TGACCCTGAAGTACCCGATAGAACATG GC-3′TAMRA.
Gene expression investigation of primary fibroblast cultures from patients with fibrotic lung disease
CCD25 lung fibroblasts were purchased from ECACC European Collection of Cell Cultures (Salisbury, UK). Fibroblasts were obtained from outgrowths of transbronchial biopsy material taken from patients with lung fibrosis at the University Hospital Freiburg (Freiburg, Germany; table 1⇓). The study received ethics approval from the appropriate hospital authorities and all patients provided informed consent. Fresh bronchial biopsies were placed on a 15-cm petridish pre-coated with collagen I (Sigma-Aldrich) in culture medium (RPMI, 1% glutamine, 1% penicillin/streptomycin and 15% foetal calf serum; Invitrogen, Karlsruhe, Germany). After 21 days, cells were trypsinised and re-cultured in 75-cm2 tissue culture flasks.
Patient information from cultured primary bronchial fibroblasts
For the fibroblast differentiation assay cells were seeded at a density of 3×105 cells. Serum-free medium was added 24 h before TGF-β2 (0.4 nM) and the inhibitors (used at concentrations of 30 nM, 100 nM, 300 nM, 1 and 3 μM). After 72 h, cells were lysed with 500 μL of Trizol (Invitrogen) and the cell lysate was stored at -80°C until further analysis.
Immunofluorescent detection of α-SMA as a marker for myofibroblasts
Fibroblasts seeded on 8-well chamber slides at a density of 5×104 cells·well−1 were incubated in serum-free RPMI medium for 24 h. Inhibitors (3 µM) were added 30 min before addition of TGF-β2 (0.4 nM). After 72 h, the medium was removed and the slides were fixed. Detection of α-SMA was performed by incubation with a monoclonal anti-α-SMA antibody (Sigma-Aldrich; diluted 1:100 with PBS) and a fluorescein isothiocyanate-conjugated rabbit anti-mouse antibody (DAKO, Glostrup, Denmark; diluted 1:500 in PBS). The slides were cover-slipped using a mixture of propidium iodide (DAKO) and mounted with Mowiol (Calbiochem).
Phospho-SMAD-2 ELISA
HaCat cells (CLS Cell Lines Service, Eppelheim, Germany) seeded into a 96-well microtitre plate at a concentration of 3×104 cells·well−1 were incubated for 2 days. Following incubation in serum-free medium for 3 h, the compounds, dissolved in medium containing 10% dimethyl sulphoxide, were added up to a final concentration of 50 µM and TGF-β1 (5 ng·mL−1) was added to the appropriate wells 15 min later. After incubation for 30 min, cells were lysed with 120 μL 10× lysis buffer (20 mM Tris-HCl; pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg·mL−1 leupeptin and 1 mM phenylmethyl sulphonyl fluoride). Lysates were stored at -80°C. To perform the phospho-SMAD-2 ELISA, a monoclonal anti-SMAD 2/3 antibody (Upstate, Dundee, UK; diluted 1:250) was coated on the surface of a 96-well plate (Nunc F8; Maxisorp, Wiesbaden, Germany) and incubated with the lysates at room temperature for 90 min. A rabbit polyclonal anti-phospho-SMAD2 antibody (Upstate) was added to the bound material and immunocomplexes were detected by addition of an alkaline phosphatase labelled mouse anti-rabbit antibody (Dako, Glostrup, Denmark) using p-Nitrophenyl Phosphate (pNPP; Upstate) as substrate. The plate was incubated in the dark at 37°C and the optical density of the signal was measured at 406 nm with an ELISA plate reader (Tecan Genios Plus; Tecan, Männedorf, Switzerland).
Determining median inhibitory values for TGF-β receptor (R)1 and TGF-βR2 kinase inhibition
The inhibitory actions of SB-431542, imatinib mesylate and BIBF 1000 on the kinase activity of TGF-βRI and TGF-βRII were determined using the Promega Kinase-GloTM kit (Promega, Mannheim, Germany) according to the manufacturer's protocol.
Statistics
All statistical comparisons were performed using unpaired nonparametric Mann–Whitney U-tests and p≤0.05 was considered to be statistically significant.
RESULTS
The effect of BIBF 1000 on the development of fibrosis in a therapeutic rat bleomycin model
BIBF 1000 (fig. 1⇓) was identified as a selective inhibitor of the family of VEGF-, PDGF- and FGF-receptor tyrosine kinases 45. To test whether BIBF 1000 would exert anti-fibrotic activity in lung fibrosis, the compound was tested in a rat bleomycin model. BIBF 1000 was used at its fully effective dose (50 mg·kg−1) in a therapeutic setting 41 with daily oral treatment from day 10–21 post-bleomycin administration. As controls, groups of rats received saline instead of bleomycin (saline group), or animals treated with bleomycin received vehicle only (bleomycin group). After 22 days, the animals were sacrificed and the level of fibrosis was determined by gene expression profiling of TGF-β1, pro-collagen-I, fibronectin and CTGF of isolated lung tissue. As shown in figure 2⇓, the gene expression of these factors is very low in the saline-treated control group and is increased after bleomycin treatment. In rats exposed to bleomycin, treatment with 50 mg·kg−1 BIBF 1000 from day 10–21 resulted in expression levels comparable to those observed in rats treated with saline alone.
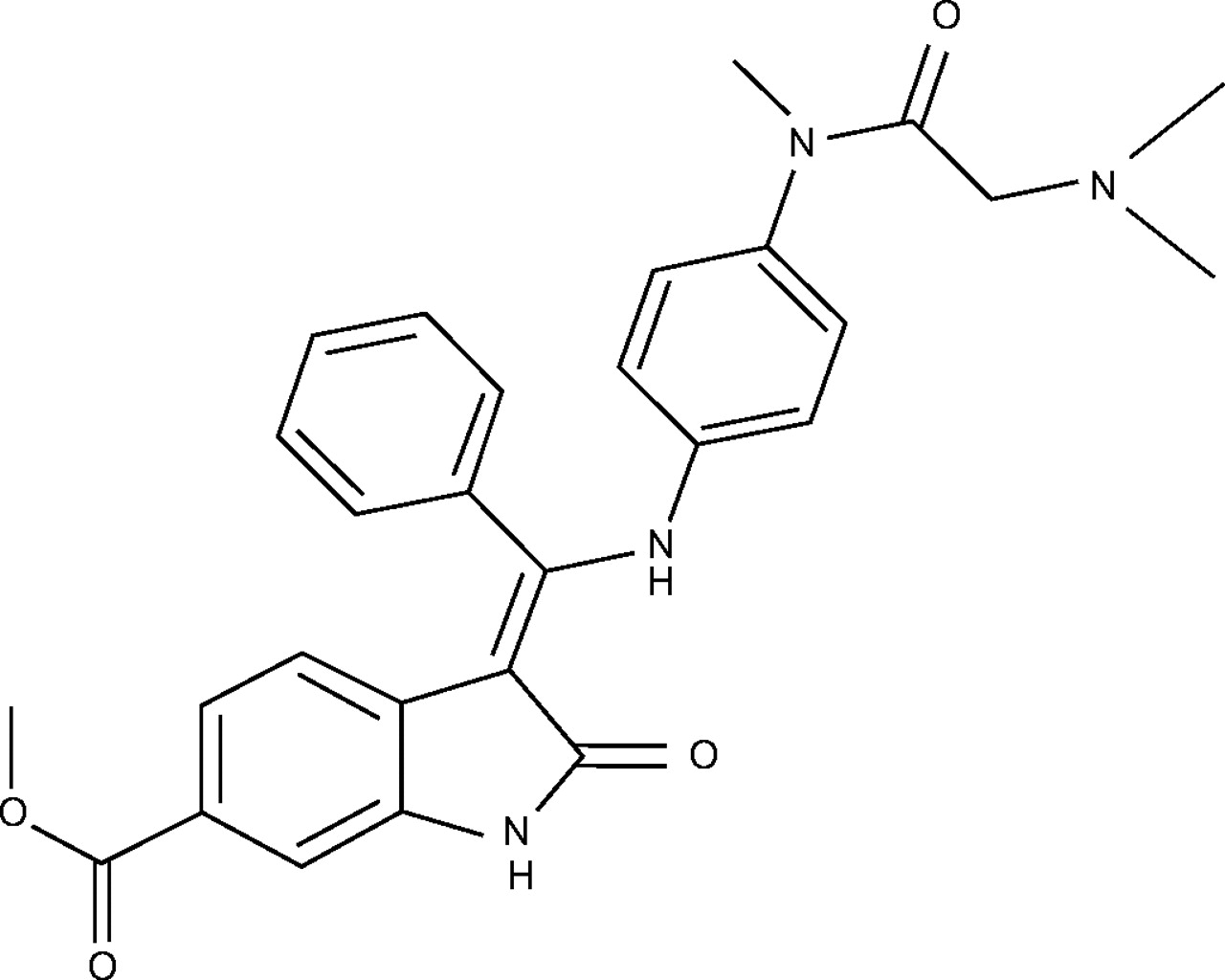
Chemical structure of BIBF 1000.

BIBF 1000 inhibits the gene expression of pro-fibrotic marker genes in the rat bleomycin (bleo) model. Rats (10 animals per group) were treated either with saline or bleo on day zero. Treatment with vehicle or BIBF 1000 (p.o. 50 mg·kg−1) commenced at day 10 and was continued daily until day 21. On day 22, rats were sacrificed and a part of the left lung lobe was processed for RNA extraction. The gene expression levels of a) transforming growth factor (TGF)-β1, b) pro-collagen I, c) fibronectin and d) connective tissue growth factor (CTGF) were determined by quantitative real-time PCR. The gene expression for each gene is indicated relative to endogenous 18S RNA control. Data are presented as fold induction and the horizontal bars represent median values. ***: p<0.001.
To address the deposition of collagens at the protein level, lung sections obtained at day 22 were stained with Massońs Trichrome. As shown in figure 3⇓, collagen deposition, as indicated by blue staining, is weak in the saline-treated control group. In contrast, rats treated with bleomycin alone showed extensive pulmonary fibrosis in the interstitial spaces. Fibrosis was strongly attenuated when bleomycin-treated rats received 50 mg·kg−1 BIBF 1000, with collagen staining levels comparable to the rats treated with saline.

Collagen staining of representative lung sections. Rats (10 animals per group) were treated either with a, d and g) saline or b, e and h) bleomycin on day zero, followed by treatment with vehicle from day 10 until day 21, or c, f and i) bleomycin on day zero, followed by treatment with BIBF 1000 (p.o.; 50 mg·kg−1) from day 10 until day 21. On day 22, rats were sacrificed and the lungs were fixed with paraformaldehyde, prior to paraffin embedding. Sections (4 μM) were stained with Masson's Trichrome stain. Muscle and cells are stained red, nuclei stained black and collagens stained blue. Three representative photomicrographs are shown for each of the groups.
TGF-β-stimulated myofibroblast formation is inhibited by BIBF 1000 in vitro
It had been previously shown that stimulation of primary fibroblasts with TGF-β induces fibroblast proliferation and differentiation into myofibroblasts 48. To determine whether BIBF 1000 would influence the TGF-β-mediated induction of myofibroblasts, primary fibroblasts obtained from outgrowths of transbronchial biopsies (table 1⇑) were treated with 0.4 nM TGF-β2 for 72 h in the absence or presence of BIBF 1000. Furthermore, SB-431542, reported to be a potent and selective inhibitor of the TGF-β superfamily of kinases 46, 49, was included as a reference. The differentiation of fibroblasts to myofibroblasts by TGF-β2 was determined by assessing the expression of α-SMA and CTGF. As shown in figures 4a⇓ and 4b⇓, cells treated for 72 h with TGF-β2 display a robust staining for α-SMA, suggesting that differentiation into myofibroblasts had taken place. In contrast, both BIBF 1000 and SB-431542 blocked the differentiation into myofibroblasts, as seen by the absence of α-SMA staining (figs 4c–4f⇓, respectively). To quantify the effects of BIBF 1000 and SB-431542, expression of α-SMA was determined by real-time PCR. As shown in figure 5⇓a and b, both BIBF 1000 and SB-431542 inhibited α-SMA gene expression (as well as CTGF gene expression, data not shown) in a concentration-dependent manner in three primary fibroblast cultures and in CCD25 lung fibroblasts.

BIBF 1000 blocks transforming growth factor (TGF)-β-mediated differentiation of fibroblasts. Fibroblasts obtained from biopsies of patients with fibrotic lung disease were cultured on collagen I coated chamber slides for 72 h in: a) serum-free medium (SFM) alone; b) SFM and 0.4 nM TGF-β2; c and d) SFM and 0.4 nM TGF-β2 and 5 μM BIBF 1000 e and f) SFM and 0.4 nM TGF-β2 and 5 μM SB-431542; or g and h) SFM and 0.4 nM TGF-β2 and 5 μM imatinib mesylate. α-Smooth muscle actin filaments (green) were detected with a monoclonal antibody and visualised with a fluorescein-conjugated rabbit anti-mouse antibody.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Activity of BIBF 1000, imatinib mesylate and SB-431542 on transforming growth factor (TGF)-β2-induced α-smooth muscle actin (SMA) gene expression and TGF-β-mediated SMAD phosphorylation. Primary fibroblast cell lines isolated from bronchial biopsies of three patients (2217: ▪; 2272: •; and 2278: ▾) with lung fibrosis and the primary lung fibroblast cell line CCD25 (▵) were incubated with TGF-β2 in the presence of a) SB-431542, b) BIBF 1000 and c) imatinib mesylate at concentrations ranging 0–30 μM. After a 72-h incubation, the gene expression levels of α-SMA were determined by quantitative real-time PCR and normalised relative to endogenous 18S RNA. Data are presented as a percentage of gene expression compared with dimethyl sulfoxide alone. HaCat cells were incubated in serum free medium with d) SB-431542, e) BIBF 1000 and f) imatinib mesylate to final concentrations ranging 0–50 μM. Data are presented as mean±SEM. After 15 min, 5 ng·mL−1 TGF-β1 was added and incubation was continued for 30 min before the cells were lysed. The amount of phosphorylated SMAD2 was determined by ELISA. A 100% value corresponds to the phosphorylation of SMAD2 after stimulation with 5 ng·mL−1 TGF-β1.
Since BIBF 1000 showed a weak inhibition of the isolated TGF-βR1 kinase (table 2⇓), it was hypothesised that cellular activities mediated by BIBF 1000 could be accounted for by direct inhibition of TGF-βR1. Therefore, a quantitative ELISA assay for the detection of phospho-SMAD2 (an immediate downstream target of TGF-βR1) was established as a marker for the intracellular activity of TGF-βRI. HaCat cells were stimulated with TGF-β for 30 min in the presence or absence of BIBF 1000 and the amount of phosphorylated SMAD2 was determined after lysis of the cells. Again, SB-431542 was used as a positive control. As shown in figure 5e⇑, treatment with BIBF 1000 at concentrations exceeding those needed to inhibit TGF-β-mediated fibroblast differentiation did not block the TGF-β-induced phosphorylation of SMAD2, whereas treatment with SB-431542 abrogated the phosphorylation of SMAD2 in a concentration-dependent manner (fig. 5d⇑). Therefore, it was surmised that BIBF 1000 blocks other cellular pathway(s) needed to induce and/or maintain the myofibroblast phenotype without directly interfering with SMAD-dependent TGF-β signalling.
Median inhibitory(IC50) values for BIBF 1000, imatinib mesylate and SB-431542 against transforming growth factor (TGF)-β receptor (R)1 and TGF-βR2
Previously, it has been shown, that imatinib mesylate exerts anti-fibrotic activity in the bleomycin-induced lung fibrosis model 40, 41. Therefore, the effects of imatinib mesylate on the TGF-β-mediated differentiation of primary fibroblasts to myofibroblasts and on the TGF-β-mediated phosphorylation of SMAD2 were examined. As shown in figures 4g, 4h⇑ and 5c⇑, imatinib mesylate did not block TGF-β-induced α-SMA expression at the protein or mRNA level in primary fibroblasts nor did it influence the TGF-β-induced phosphorylation of SMAD2 in HaCat cells (fig. 5f⇑).
DISCUSSION
The use of different treatment regimes in the bleomycin model may prove a valuable method by which drugs with true anti-fibrotic potential can be identified and investigated 40, 41. The present study tested BIBF 1000, previously identified as an inhibitor of the receptor tyrosine kinases for VEGF, FGF and PDGF, and demonstrated that BIBF 1000 attenuates established lung fibrosis in an in vivo setting. Furthermore, this compound blocked TGF-β-mediated differentiation of human primary lung fibroblasts isolated from lung fibrosis patients.
Inhibition of the pathways regulated by CTGF, IGF-1, VEGF, FGF, PDGF and TGF-β have been suggested to provide novel therapeutic approaches for the treatment of fibrosis associated with chronic lung diseases. As discussed earlier, each of these growth factors has distinctive roles in the pathophysiology of fibrosis and many are induced by TGF-β. However, the relative contribution of each of these pathways for the pathogenesis of lung fibrosis remains obscure and may depend on the specific stage and type of the fibrotic disease. TGF-β is the most potent pro-fibrotic growth factor known and it has been shown that interference with the TGF-β-pathway will attenuate fibrosis of different origin 29, 50–52. However, direct inhibition of TGF-β-signalling, e.g. via small-molecule inhibition of TGF-β receptor kinases, may not offer a viable therapeutic option due to the pleiotropic functions of this growth factor, which suggest that a number of side-effects, including especially SMAD-dependent promotion of tumour formation, might be associated with a long-term anti-TGF-β-treatment 31, 32. These concerns are particularly important in light of the dramatically increased lung cancer rates seen in IPF patients 53, 54. Therefore, it was interesting to note that BIBF 1000 was able to block TGF-β-mediated differentation of primary fibroblasts isolated from normal lungs and from patients with fibrotic lung diseases in the absence of inhibition of the TGF-β receptor kinases. This suggests that fibroblasts transform to myofibroblasts through the actions of TGF-β via downstream factor(s) which are inhibited by BIBF 1000. Since differentiation of fibroblasts to myofibroblasts is a phenomenon seen in fibroblasts isolated from the normal lung and from a number of different diseases, including asthma 13, 55, liver cirrhosis 56, renal fibrosis 57, sarcoidosis, IPF and usual interstitial pneumonia, BIBF 1000 or related compounds may be of general utility in a number of fibrotic diseases.
It has been shown that c-abl is a SMAD-independent signalling molecule downstream of TGF-β required for morphological transformation and expression of ECM 58. Although the current authors and others have previously shown that imatinib mesylate (a PDGF receptor/c-abl/c-kit inhibitor) is efficacious in the bleomycin-induced lung fibrosis model 40, 41, 58, little effect on the differentiation of fibroblasts was observed following treatment with imatinib mesylate, indicating that neither PDGF nor c-abl (nor their combination) are the sole mediators of the differentiation process. As shown by global expression profiling 59, >100 genes play a role in TGF-β-mediated fibroblast-myofibroblast differentiation. Future cell culture experiments comparing gene expression profiles with the inhibitors described in the present study could provide important clues about the mechanism of TGF-β-mediated fibroblast-myofibroblast differentiation.
Since BIBF 1000 is an inhibitor of the receptor tyrosine kinases for PDGF, FGF and VEGF, it is tempting to speculate that the concerted inhibition of several pro-fibrotic factors is required for its anti-fibrotic activity. PDGF is believed to play a role in the pathogenesis of fibrotic disease by stimulating fibroblast chemotaxis, proliferation and fibroblast-mediated matrix contraction 60. Furthermore, PDGF is important in inducing the secretion of growth factors and ECM components in fibroblasts 19, inducing fibroblast proliferation and the production of fibronectin by both normal and fibrotic lung fibroblasts. Interestingly, PDGF did not have any effect on the production of interstitial collagens, again, supporting the hypothesis that the concerted action of several factors may be required to induce all aspects of fibrosis. bFGF or FGF-2 is released by activated fibroblasts and damaged epithelial cells during remodelling processes associated with bronchial asthma 14, 61–63 and it stimulates the proliferation and fibronectin production of human lung fibroblasts. Furthermore, TGF-β1-induced proliferation of fibroblasts is mediated through the release of extracellular FGF-2 since FGF-2-blocking antibodies inhibited the proliferation of fibroblasts 19, 61. Finally, it has been shown that both PDGF and FGF-2 are important factors in the migration of myofibroblasts 64, suggesting that blockade of both pathways might be required to interfere with myofibroblasts.
The function of angiogenesis and of pro-angiogenic factors like VEGF for the pathophysiology of pulmonary fibrosis is currently not understood. Neovascularisation with anastamoses between the systemic and pulmonary vasculature is apparent at sites of fibrosis 44, 65. However, a regional heterogeneity of the vascularisation in IPF patients has been reported and it has been proposed that this heterogeneity may at one site support fibroproliferation but at other sites may block the normal repair mechanisms 65. Although the exact site and mechanism of the neovascularisation remains controversial, it is tempting to speculate that angiogenesis may play a role in IPF, ARDS and other lung fibroses, and that the use of VEGF inhibitors might attenuate these processes.
The current authors presume that the combined VEGF receptor, FGF receptor and PDGF receptor inhibition of BIBF 1000 is acting in a concerted manner to control fibrosis. Of course the possibility that inhibitory effects of some as yet unidentified targets of BIBF 1000 may also play a role in this process cannot be ruled out.
CONCLUSION
In summary, the present data suggest that BIBF 1000, or a molecule with a similar kinase inhibition profile, may present a novel therapeutic opportunity with which to treat interstitial pulmonary fibrosis. Its distinctive inhibitory profile is uniquely capable of preventing fibroblast-myofibroblast differentiation, a crucial step in the establishment of fibrosis, without directly affecting SMAD signalling. Ultimately, only clinical trials in interstitial pulmonary fibrosis and other fibrotic diseases will show whether such compounds can stop or slow the inexorable course of this invariably fatal disease.
Statement of interest
None declared.
Acknowledgments
The excellent technical assistance of E. Mueller, S. Mueller, M. Ried and M. Trojan (all Dept of Pulmonary Research, Boehringer Ingelheim Pharma GmbH&Co. KG, Biberachan der Riss, Germany) is acknowledged.
- Received November 23, 2006.
- Accepted January 19, 2007.
- © ERS Journals Ltd
References










