





Abstract
Genetic studies in familial lung fibrosis have demonstrated an association with surfactant protein C genes: two mutations have been found resulting in protein misfolding and causing type-II epithelial cell injury. Remarkably, different histological patterns were observed in the affected subjects, suggesting the influence of modifier genes and/or environmental factors. Surfactant protein C gene variations have not, however, been associated with sporadic cases, i.e. idiopathic pulmonary fibrosis (IPF).
Susceptibility to IPF probably involves a combination of polymorphisms related to epithelial cell injury and abnormal wound healing. To date, the genetic associations with IPF that have been reported in different cohorts include the genes encoding tumour necrosis factor (TNF; −308 adenine), interleukin-1 receptor antagonist (+2018 thymidine) and association with severity and progression (interleukin-6/TNF receptor II and transforming growth factor-β1 (TGFB1; +869 cytosine)), but none of these associations have been replicated by others.
Unlike in IPF, immunological inflammation seems to be more prominent in the pathogenesis of scleroderma lung fibrosis, being an autoimmune disease with specific autoantibodies, such as antitopoisomerase antibodies, in patients with diffuse lung disease, and anticentromere antibodies, in patients with pulmonary vascular disease. Antitopoisomerase antibody positivity is associated with the carriage of human leukocyte antigen DRB1*11 and DPB1*1301 alleles, suggesting the recognition of a specific amino-acid motif. Extended haplotype analysis also supports the conclusion that TNF may be the primary association with anticentromere positivity. Intriguingly, associations with TGFB1 and genes involved in extracellular matrix homeostasis have been reported in this disease.
In conclusion, significant steps forward have been taken in the understanding of the genetic contribution to fibrosing lung diseases, but major challenges lay ahead. It is the present authors' opinion that only a combined approach studying large numbers of familial and sporadic cases, all clinically well phenotyped, using multiple distinct cohorts, and genotyped according to relevant gene ontologies will be successful. It will be necessary to be particularly vigilant with regard to phenotype; the absence of very strong reproducible associations may be because of the rigidity of phenotype definition, coupled with the possibility that idiopathic pulmonary fibrosis may still be a heterogeneous group of diseases, despite the more rigid definition set out by the European Respiratory Society/American Thoracic Society statement.
Diffuse parenchymal lung diseases (DPLDs) comprise >200 entities and include a wide spectrum of diseases, many uncommon and of unknown aetiology, but together accounting for ∼15% of respiratory practice 1. An important heterogeneous group of DPLDs are the idiopathic interstitial pneumonias (IIPs), which result from damage to the lung parenchyma by varying mechanisms of inflammation and fibrosis. Recently, an international statement on IIPs has been published that, for the first time, provides an integrated clinical, radiological and pathological approach to the classification of these diseases, including seven clinicoradiopathological entities 2. Idiopathic pulmonary fibrosis (IPF) is the best-known entity and can now be diagnosed even without the need for surgical lung biopsy. Nonspecific interstitial pneumonia (NSIP), cryptogenic organising pneumonia, acute interstitial pneumonia, respiratory bronchiolitis-associated interstitial lung disease, desquamative interstitial pneumonia and lymphoid interstitial pneumonia are the other disease entities 2. Importantly, radiological and histological patterns seen in IIPs can also be found in the context of systemic diseases, or exposure to drugs or other environmental factors. For example, NSIP is the most common DPLD in patients with systemic sclerosis.
It is widely believed that genetic factors play an important role in the aetiopathogenesis of many DPLDs, but the identification and understanding of these factors is in its infancy. This review focuses on key issues in the genetic research of fibrosing lung diseases, and, in particular, summarises current knowledge about genetic predisposition to familial pulmonary fibrosis, sporadic IPF and lung fibrosis in systemic sclerosis.
KEY CONSIDERATIONS
Many DPLDs can result in “pulmonary fibrosis”. Historically, (idiopathic) interstitial pneumonias were often considered as being synonymous with “pulmonary fibrosis”, and inappropriate attempts were made to derive meaningful correlations of treatment response and outcome in this population. Additionally, lung fibrosis can result from many other pathological processes, including infection, malignancy and surgical procedures. Thus, the terms “lung/pulmonary fibrosis” and also “fibrosing alveolitis” should not be used as a diagnostic label. Recent refinement in the classification of IIPs has provided an important handle for diagnostic categorisation of fibrosing lung diseases 2. Even within these diagnostic categories, considerable heterogeneity in terms of clinical characteristics, disease activity, severity, prognosis and response to therapy can be observed. Therefore, in the context of genetic studies, precise phenotyping of each case is extremely important.
It is thought that the development of most fibrosing lung diseases occurs in susceptible individuals following an exposure to a variety of potential environmental triggers. Evidence to support this hypothesis derives from the observation of varying susceptibility to environmental causes; reports on familial clustering of DPLDs, including IPF, sarcoidosis, hypersensitivity pneumonitis, Langerhans' cell histiocytosis, pulmonary alveolar proteinosis and desquamative interstitial pneumonia; and from human leukocyte antigen (HLA) association studies.
Fibrosing lung diseases are referred to as “complex diseases”, i.e. it is thought that multiple genetic loci, each exerting variable relatively small effects, are involved. Some alleles might predispose to disease, whereas others might be protective (susceptibility alleles), or might be involved in severity/progression (modifying genes).
GENETIC RESEARCH STRATEGIES
Three main strategies can be applied to investigation of the role of genetic factors in fibrosing lung diseases. These strategies do not differ from those used in other fields.
The first strategy is linkage analysis in families. Linkage analysis requires the use of a large number of DNA markers that are closely spaced throughout the genome in order to attempt to identify chromosomal regions that cosegregate with the occurrence of disease or a specific phenotype in families with lung fibrosis. Once a candidate region is sufficiently small, the disease-associated gene can be located through positional cloning if there are no other candidate genes in this area. There are advantages and disadvantages to such a classic linkage analysis. An advantage is that a priori no candidate gene has to be defined. The definition/selection of the latter is often an educated guess or based on insight. A clear disadvantage is the preferable need for three generations: DNA from grandparents, parents and children are needed, and, when the disease occurs late in life, grandparental information is often lacking. A second problem with these studies is the need for substantial sample sizes.
The second strategy is based on case–control association studies, which seek evidence for a significant association between an allele of a candidate gene or gene haplotype (series of polymorphisms in the same gene that cosegregate) and a disease/phenotype characteristic by comparing the allele/genotype frequencies of affected subjects and carefully selected and, if necessary, age-matched controls. There is no need for information derived from grandparents or children and, hence, this strategy is often the preferred route for late-onset diseases. Case–control studies are relatively easy to perform in a short period of time, which is one of the reasons why they have became increasingly popular. Case–control studies, however, have the problem that a significant association between an allele and a disease does not mean that the candidate gene is a causal one. Population stratification in the (distant) past can lead to allele frequency differences between diseased and healthy subjects without the gene being causative or disease related. A positive finding in a case–control study, therefore, always needs confirmation in another group of subjects whom it might reasonably be assumed are not related to the first group of subjects, e.g. a positive finding in Japanese subjects should be confirmed in subjects who are African, Dutch, English or of some other race before that allele can be added to the list of “approved” candidate genes. The obvious need for confirmatory studies may render the case–control study less attractive.
Thirdly, the affected sibling pair (ASP) method might be used. This studies identity by descent sharing between two siblings affected by the disease of interest. Basically, the ASP test compares the frequencies of alleles in diseased and nondiseased subjects and shares many of the characteristics of the case–control study, the difference being that related subjects (siblings) now form the control group. The statistical evaluation of such correlated information is more demanding because the straightforward Chi-squared test cannot be used: specialised statistical software must be used. The major advantage of the ASP test is, however, that population stratification is no longer a problem: it starts from the notion that, if population stratification does exist, the siblings of the diseased subject will be affected by that process to the same degree, and so any difference in allele frequency between diseased and nondiseased subjects must reflect a genuine association.
FAMILIAL PULMONARY FIBROSIS
Familial pulmonary fibrosis or familial IIP is identified by confirming IIP in two or more members of the same family. It is of note that different subtypes of IIP can be found in an affected family, which sometimes causes difficulties in establishing a precise familial disease phenotype. The most frequently reported IIP entity in familial-type lung fibrosis is IPF/usual interstitial pneumonia (UIP; ∼80%) 3.
Epidemiological studies
The precise incidence and prevalence of IPF are not known. Its incidence has been estimated at 7–11 per 100,000 in the general population 4. The prevalence has been estimated at 13–20 per 100,000 population and increases with the ageing process 4. Marshall et al. 5 were the first to report a large cohort of familial IIP. They identified 25 UK families, comprising 67 cases, which were all classified as IPF on the basis of high-resolution computed tomographic findings (93%) and/or the histological pattern (32%). The authors estimated that familial cases account for 0.5–2.2% of all patients with IPF, with a prevalence of 1.34 cases per million in the UK population. In contrast, Loyd 6 recently suggested that the real incidence of familial IPF might be considerably higher. The author reported that, in the Vanderbilt lung transplantation programme, nine of the 47 (19%) individuals undergoing transplantation for this condition showed a positive family history.
In a nationwide epidemiological study in Finland including all pulmonary clinics in the country (n = 29), a prevalence of 5.9 per million population was found for familial IPF 7. Interestingly, this study revealed evidence of geographical clustering of multiplex families, suggesting a recent founder effect in patients with familial IPF 7.
In the largest familial study undertaken to date, coordinated by D. Schwartz in the USA, 75 families with two or more affected individuals have been identified. In 56 of these, detailed phenotyping has shown that 25 families exhibited uniform IPF, whereas, in the remaining 31, phenotypic heterogeneity was observed. The group is undertaking detailed linkage analysis in an endeavour to find a region sufficiently tight for positional cloning and hopefully to identify a key gene or genes (D. Schwartz, Duke University Medical Center, Durham, NC, USA, personal communication).
Mode of inheritance
The familial form of IPF/UIP is probably transmitted as an autosomal dominant trait with reduced penetrance 3, 8, 9. An interesting observation in this respect has been made by Bitterman et al. 9. They evaluated 17 unaffected members of three families with an autosomal dominant form of IPF and found evidence of alveolar inflammation in approximately half of them. In addition, these subjects showed no progression towards lung fibrosis during a follow-up of 2–4 yrs. Unfortunately, to the present authors' knowledge, longer-term follow-up data from this study cohort have not been reported.
Surfactant protein C gene mutations
As the results of genetic studies analysing candidate loci near the HLA region of chromosome 6 and on chromosome 14 in familial lung fibrosis have remained largely negative 6, 10, clearly one of the most intriguing genetic findings to date in this disease has been the identification of causal mutations in the surfactant protein gene (SFTP)C. Nogee et al. 11 were the first to describe a mutation in this gene that was associated with NSIP in an infant whose mother had desquamative interstitial pneumonia. Heterozygous guanine (G) to adenine (A) transition substitution (>) of the first base of intron 4 (IVS4+1G>A) was present in both patients, and caused skipping of exon 4 with the deletion of 37 amino acids. The putatively encoded product thus lacked a cysteine residue that is important for disulphide-mediated protein folding. In the patients, lack of mature surfactant protein (SP)-C in lung tissue and bronchoalveolar lavage fluid was observed, supporting the concept that the precursor protein was not being processed and secreted normally. The SFTPC IVS4+1G>A mutation was identified on only one allele in each patient, consistent with an autosomal dominant pattern 11. Recently, transfection studies demonstrated that the mutant protein diverts its wild-type counterpart to aggresomes, thus providing a molecular mechanism for the dominant negative effect observed in vivo 12.
Consistent with this finding, Thomas et al. 8 reported on another mutation in the gene encoding the hydrophobic lung-specific SP-C. In a large familial IIP kindred, including 14 affected members and spanning six decades, they found a thymidine (T) to A transversion in exon 5 (position +128) in DNA from all available affected family members. This mutation substituted a polar residue (glutamine) for a highly conserved neutral amino acid (leucine 188) that was predicted to hinder processing of SP-C precursor protein. This was confirmed by immunostaining for pro-SP-C, which showed very abnormal distribution of staining in lung from affected patients in this family compared with normal lung 8. Interestingly, again, two different pathological diagnoses (UIP and NSIP) in affected relatives sharing the same SFTPC mutation were found. In addition, the histological diagnosis varied with age, i.e. UIP in adulthood and NSIP in childhood. Although the possibility of NSIP occurring as precursor lesion to UIP could not be excluded in this family, it is more likely that the pleiotropic effects of the genetic defect are caused by modifier genes and/or environmental factors.
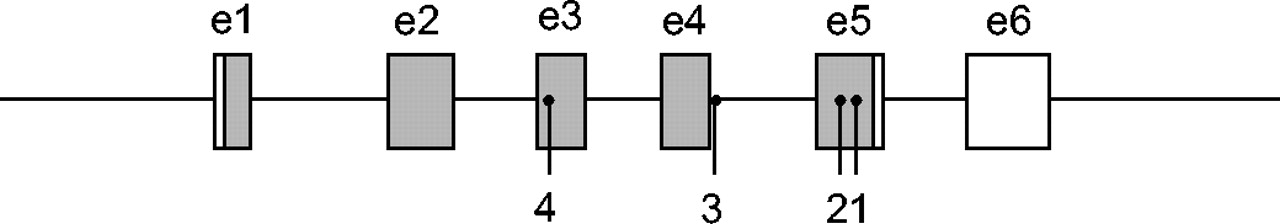
SFTPC has been localised on the short arm of chromosome 8 13. It is a relatively small gene spanning 3.5 kilobases and composed of six exons 14, with expression restricted to type-II alveolar cells. Interestingly, two other mutations in SFTPC (exons 3 and 5) have recently been reported in infantile pulmonary alveolar proteinosis with or without interstitial lung disease 15, 16. Figure 1⇓ shows a diagram of the SFTPC gene with the nucleotide positions of the previously described mutations.
{kind=link}
Nucleotides 22,041,255–22,043,927 of chromosome 8, showing the current nucleotide positions (according to Riva and Kohane 17) of the surfactant protein C gene mutations currently known to be associated with interstitial lung diseases: 1) 22,043,461T>A (glutamine substituted for leucine 188) 8; 2) 22,043,398 G>A (glutamine substituted for arginine 167) 16; 3) 22,042,998 G>A (skipping of exon 4) 11; and 4) 22,042,547T>C (threonine substituted for isoleucine 73) 15, 16 (□: noncoding region; ▓: coding region). Mutations 1 and 3 have been attributed to the occurrence of pulmonary fibrosis in different families 8, 11. e: exon.
Proposed surfactant protein C gene mutation disease mechanisms
SP-C is perhaps the most hydrophobic protein yet encountered in the mammalian proteome, bearing a domain rich in valine, leucine and isoleucine residues that form a stable α-helical structure approximately spanning the phospholipid bilayer 18. The α-helical SP-C molecule is membrane associated and palmitoylated, and readily forms insoluble random structures in the aqueous environment 19. Missense or short deletion mutations, as seen in the studies of Nogee et al. 11 and Thomas et al. 8, result in the production of a stable mRNA that produces an abundance of a misfolded protein, which, by accumulation or complex formation, may cause type-II epithelial cell injury. However, Amin et al. 20 described a family with similar lung pathology in whom expression of pro-SP-C and active SP-C was undetectable. Therefore, the severe lung disease caused by abnormalities in SP-C can result from production and accumulation of an abnormal pro-SP-C as well as its absence. A deficiency of SP-C in surfactant could cause abnormal shear forces in the alveoli, thereby causing mechanical injury of the respiratory epithelium, which, in turn, may contribute to the pathogenesis of (familial) pulmonary fibrosis 6.
Hermansky–Pudlak syndrome gene mutations
Hermansky–Pudlak syndrome (HPS) is an autosomal recessive disorder characterised by the classic triad of oculocutaneous albinism, bleeding and lysosomal ceroid (a chromolipid related to lipofuscin) storage resulting from defects in multiple cytoplasmic organelles: melanosomes, platelet-dense granules, and lysosomes 21. A certain fraction of HPS patients also suffer from pulmonary fibrosis, which is often fatal before 40 yrs of age. Although the disease occurs worldwide, it is most common in north-western Puerto Rico, where its frequency is 1 in 1,800 owing to a founder effect. In Europe, the disease has also been identified in a village in the Swiss Alps, and in persons of Dutch and Turkish ethnicity. Four disease phenotypes are currently recognised, i.e. HPS-1, HPS-2, HPS-3 and HPS-4, of which HPS-1 and HPS-4 are associated with severe pulmonary fibrosis. The HPS phenotypes are recognised to result from mutations in four genes: HPS-1 gene, adaptin β3A gene (ADTB3A), HPS-3 gene, and HPS-4 gene, respectively 21. To date, only ADTB3A produces a product of known function. This gene codes for the β3A subunit of the adapter complex-3 coat protein, a heterotetrameric complex that mediates vesicle formation 22, 23. Although no direct evidence exists to support the hypothesis, it would be logical that the other HPS-causing genes are also involved in vesicle formation.
The pathological features of HPS lung fibrosis have been studied by Nakatani et al. 24, and are characterised by extensive proliferation of type-II epithelial cells with characteristic foamy swelling/degeneration, patchy fibrosis with lymphocytic and histiocytic infiltration, and honeycomb change. Unlike in UIP/IPF, the honeycombing was not found to show a predilection for the lower lobes or subpleural regions. Type-II epithelial cells are characterised by an accumulation of phospholipid, with weak positivity for surfactant protein immunohistochemically. Ultrastructurally, the presence of numerous giant lamellar bodies that compress the nucleus with occasional cytoplasmic disruption has been demonstrated 24. Together these findings suggest a form of cellular degeneration with an accumulation of surfactant (giant lamellar body degeneration), indicating that there might be a basic defect in the vesicle formation/secretion process of surfactant by type-II epithelial cells in HPS.
SPORADIC IDIOPATHIC PULMONARY FIBROSIS
Although SFTPC mutations form a genetic basis for some familial cases of IPF, they are not associated with all of the families with IPF. In addition, the majority of patients with IPF lack a family history. Therefore, the development of IPF is likely to be determined by multiple genetic factors that each contributes to a modest effect on predisposition to this disease. In combination with appropriate environmental or cellular triggers, individuals who possess these predispositional factors may develop IPF. To date, the lack of genome-wide linkage analysis studies and knowledge of chromosomal loci for IPF make the positional cloning of these genes impossible. Therefore, case–control association studies are presently the method of choice for the genetic study of IPF.
Following the traditional paradigm for the pathogenesis of IPF, candidate genes that have been analysed for this disease are primarily involved in inflammatory responses, e.g. tumour necrosis factor (TNF)-α and genes in the interleukin (IL)-1 gene cluster. However, increasing evidence since the late 1990s suggests that the inflammatory response may not precede the fibrosis nor play a major role in the development of IPF 25. Instead, the evolving hypothesis proposes that IPF is a disease of impaired wound healing involving the epithelial/fibroblast pathway 26. The triggering event in IPF is thought to be multiple and continuous microscopic insults to the alveolar epithelial cells, suggesting that different gene ontologies might be relevant. Specifically, candidate genes might be found in surfactant genes and genes involved in oxidant/antioxidant mechanisms. Epithelial injury is followed by activation of the coagulation cascade, and aberrant wound healing subsequently leads to fibroblast proliferation, transformation into myofibroblasts, formation of fibroblastic foci, deposition of extracellular matrix and, ultimately, end-stage fibrosis. It is obvious from this extensive pathogenetic process that many candidate genes can be identified with reasonable logic. In that the insult to the epithelial cell is probably the first event in this process, genes involved in the epithelial cell response to this insult might be considered as the most attractive targets, but the scientific literature is very light in this regard.
Although many candidate genes for a key role in IPF pathogenesis can be proposed, only limited numbers have been evaluated to date, and even fewer of them have demonstrated confirmed associations. Candidate genes and polymorphisms can best be categorised according to specific biological domains playing a role in IPF pathogenesis. Each major category will be discussed, including a review of the results reported to date, which are also summarised in table 1⇓. It is of note that, because of the intrinsic pitfalls of genetic association studies 27, and the chances of spurious classification of phenotypes and insufficiently powered case–control studies, positive and negative findings should be interpreted with caution. Positive findings, suggesting putative disease gene variants in the development of disease, need further replication and, ultimately, the demonstration of a biologically functional effect. Negative findings do not rule out a gene when only a single nucleotide polymorphism has been studied. Before a candidate gene can be confidently removed from the list, detailed knowledge of linkage disequilibrium across the gene is regarded as a prerequisite.
Genes and gene polymorphisms that have been evaluated in sporadic idiopathic pulmonary fibrosis
Pro-/anti-inflammation
Interleukin-1 gene cluster
IL-1 comprises two structurally distinct forms, IL-1α and IL-1β. Both are potent proinflammatory cytokines with fibrogenic properties. The genes encoding IL-1α (IL1A) and IL-1β (IL1B), and their naturally occurring inhibitor IL-1 receptor antagonist (IL1RN), are localised in a cluster on chromosome 2q14. Whyte et al. 30 have demonstrated an association between IL1RN and IPF, i.e. the rarer allele of the +2018C>T polymorphism conferred an increased risk of IPF in subjects from the UK and Italy. However, an IL1RN association with susceptibility to IPF could not be confirmed by others 29. The same investigators did not find an association for multiple polymorphisms in IL1A and IL1B either 29. However, an association between the −889T allele of IL1A and severity of gas transfer deficits in patients with IPF has been reported 28.
Tumour necrosis factor and related genes
Expression analysis of TNF-α in lung tissue from patients with IPF has shown elevated levels in (regenerating) type-II pneumocytes 41–43. Whyte et al. 30 first reported an association between the TNF gene and IPF. Carriage of the TNF −308A allele was associated with increased risk of IPF in well-defined UK (odds ratio (OR) 1.85) and Italian (OR 2.50) cohorts 30. Riha et al. 31 recently confirmed this association in 22 Australian IPF patients. However, a study by Pantelidis et al. 32 did not detect an association between the TNF −308A allele and IPF in UK patients. In addition, no association was found for multiple other polymorphisms in the TNF gene, and polymorphisms in the genes encoding TNF receptor II (TNFRII) and lymphotoxin-α 34. Interestingly, the G allele of the IL6 intron 4 A>G polymorphism was associated with lower carbon monoxide diffusing capacity of the lung, and co-carriage of the TNFRII 1690C allele and the IL6 intron 4G allele was associated with presence of disease, suggesting a combinatory effect of these two genes in IPF development 32.
Cytokines considered to have an anti-inflammatory effect, for example IL-10, have a regulatory role affecting TNF-α production. Alveolar macrophages from patients with IPF exhibit elevated levels of TNF-α relative to IL-10, suggesting that, in this disease, the normal homeostatic mechanisms fail to regulate the TNF-α inflammatory response 33. There is evidence that IL-10 gene haplotypes and promoter polymorphisms are associated with differential IL-10 production, and ∼75% of interindividual variation in IL-10 production appears to be genetic 44, 45. Recently, Whittington et al. 33 screened the coding sequence and 3' untranslated region of IL10 for polymorphisms in 96 patients with IPF. Although they identified a novel polymorphism at nucleotide position +43 from the start (G>A), causing an amino acid change and resulting in lower levels of IL-10 secretion, no difference in allele frequency between patients and controls was observed.
Chemokine genes
Neutrophil accumulation in the lower respiratory tract is a typical finding in IPF, and the recruitment and activation of neutrophils is believed to play a fundamental role in the development of lung injury that precedes normal repair. IL-8 has been found at increased levels in the lungs of patients with IPF, and several studies have shown a correlation between IL-8 levels in bronchoalveolar lavage fluid or expression of alveolar macrophage mRNA IL-8 and bronchoalveolar lavage fluid neutrophil count in this disease. Only one study has investigated single-nucleotide polymorphisms in the genes encoding IL-8 and its receptor, but no association was found 34.
Genes involved in T-helper cell type 1/type 2 response
Ample data support the role of T-helper cell type 2 regulatory cytokines in the pathogenesis of lung fibrosis 46. This type of cytokine response is associated with high levels of IL-4, -5 and -13, and a paucity of interferon (IFN)-γ 47–49. IFN-γ shows antifibrotic properties, and this imbalance might thus contribute to the excessive fibroblast activation, deposition of collagen and scar formation that occurs in IPF. Functional polymorphisms in genes encoding cytokines influencing T-helper cell type 1/type 2 balance, and especially IFN-γ and its receptors, have, therefore, been studied in IPF.
IL-12 plays a key role in inducing IFN-γ production, and a single nucleotide polymorphism in the 3' untranslated region of the IL12 p40 gene (i.e. IL12B) at position 1188 has been shown to correlate with increased IL-12 secretion 50. Latsi et al. 35 found no association of this polymorphism with susceptibility to IPF. Similarly, they found equal distribution of a potentially functional polymorphism in the IFN-γ gene (position 5644 in the 3' untranslated region) in IPF and control subjects. However, absence of an association with just a single nucleotide polymorphism does not exclude a role for this gene in genetic susceptibility to IPF.
Complement receptor genes
Complement receptor 1 (CR1/CD35/C3b/C4b receptor) is important in the clearance of immune complexes. The C>G polymorphism at nucleotide position +5507 in exon 33 has been correlated with the levels of CR1 expressed on erythrocytes, which may directly affect the clearance of immune complexes 36. Although there is no firm evidence for a role of immune complex clearance in the pathogenesis of IPF, this is an attractive concept. Zorzetto et al. 36 analysed this and two other polymorphic sites in the CR1 gene using an Italian IPF cohort. They demonstrated a significant association between the GG genotype of the C5507G polymorphism and IPF (OR 6.2) 36. As this GG genotype is thought to result in low expression of CR1 on erythrocytes, the authors speculated that, in a subset of subjects who develop IPF, CR1 polymorphisms related to a low CR1/erythrocyte ratio might contribute to impaired clearance of immune complexes containing viral particles and/or complement-opsonised viruses. In conjunction with other environmental and genetically determined factors, this could result in repeated episodes of lung injury and subsequent aberrant wound healing.
Pulmonary surfactant
Association of mutations in SFTPC with familial IPF suggests that genes involved in maintaining the alveolar structure and function are important for the development of IPF. In addition to SFTPC, SFTPA1, SFTPA2, SFTPB and SFTPD are also members of the surfactant protein gene family 51. Using a Mexican IPF cohort, Selman et al. 37 evaluated gene polymorphisms in SP-A1, SP-A2, SP-B, SP-C and SP-D genes. Stratification of the study cohort based on their smoking habit demonstrated associations for the 6A4 haplotype of SFTPA1 with smoking IPF patients, and for the 1580C allele of SFTPB with smoking IPF patients. Functional analyses of the SP-A with different SFTPA1 haplotypes demonstrated 6A4-specific aggregation of SP-A. It is of note that the SFTPB 1580C allele has also been shown to confer risk of chronic obstructive pulmonary disease, making a specific role in IPF pathogenesis unlikely 52. Furthermore, associations with sporadic IPF cases were not detected for multiple polymorphisms in SFTPC and SFTPD 37.
More recently, Lawson et al. 53 reported SFTPC sequence results from 89 patients with sporadic IPF and found evidence for a role of genetic mutations in only 1% of cases. These findings may be in line with the results of Amin et al. 20, who demonstrated preserved SP-C expression in bronchoalveolar lavage fluid and lung biopsy specimens from 19 unrelated IPF patients. Therefore, it is unlikely that SFTPC mutations contribute to the pathogenesis of IPF in the majority of sporadic cases.
Oxidative/antioxidative balance
Several studies have shown excessive oxidant production in IPF, as well as deficiencies in glutathione production 54, 55. Increased production of oxidants has been suggested to contribute to epithelial cell apoptosis, which is an important early feature in the pathogenesis of pulmonary fibrosis. Loss of the alveolar epithelial protective barrier could subsequently lead to exposure of the underlying basement membrane to oxidative injury, resulting in degradation of key constituents of basement membrane. Loss of the integrity of the subepithelial basement membrane is commonly thought to be an essential and unique element in the progressive pulmonary fibrosis of IPF 56. Therefore, genes involved in oxidative/antioxidative balances can be regarded as important candidates for genetic studies in IPF. However, to date, no such studies have been reported.
Coagulation cascade
Increased local procoagulant and antifibrinolytic activities have been found in patients with IPF, suggesting that fibrin matrix removal is reduced, and, as a consequence, fibroblast migration, extracellular matrix deposition and the magnitude of the fibrotic response are increased 57, 58. Levels of tissue factor and plasminogen activator inhibitor (PAI)-1 and PAI-2 are significantly elevated in bronchoalveolar lavage fluid obtained from IPF patients, and the compensatory increase in tissue factor pathway inhibitor appears to be insufficient to counterbalance tissue factor, which leads to a hypercoagulable state in IPF lungs 59. Kim et al. 38 have recently evaluated a polymorphism in the PAI-1 gene, but did not find an association with IIP. However, further systematic analysis of these genes using functional genomic tools is warranted to determine their roles in the development of IPF.
Fibroblast-related pathways
Transforming growth factor-β
Fibroblast migration/proliferation and phenotypic change to myofibroblasts, with subsequent accumulation and remodelling of extracellular matrix are central to the pathogenesis of IPF 25, 46. Transforming growth factor (TGF)-β1 has been shown to be a critical mediator of lung fibrosis in animal models, and a number of studies have shown that antagonising TGF-β1 prevents the development of tissue fibrosis 60. Furthermore, targeted overexpression of TGF-β1 has been shown to produce progressive fibrosis 61. Interestingly, recent insights from rat models show that TGF-β3 is associated with “regular” wound healing, whereas TGF-β1 is associated with fibrotic wound healing 62. TGF-β3, therefore, seems to acts as an endogenous counterbalance of TGF-β1.
There is evidence that the circulating concentration of TGF-β1 is under predominantly genetic control 63, implying that genetic variation at the TGF-β1 gene (TGFB1) locus might influence diseases, including IPF, in which TGF-β1 is implicated. Against this background, Xaubet et al. 39 have assessed polymorphisms in TGFB1 in White IPF patients from Spain. They studied two exon 1 polymorphisms, at positions +869 (T>C) and +915 (G>C), both resulting in amino acid substitutions. The authors found no association of either polymorphism with susceptibility to IPF. Interestingly, however, the +869C allele (codon 10 proline) was associated with increased deterioration of the disease, as measured by gas exchange 39. Although this allele was not associated with other functional parameters of IPF, and the levels of TGF-β in both case and control subjects were not determined, the study provides the first evidence for TGFB1 as a potential determinant of disease progression in IPF. However, further evaluation using independent IPF cohorts is necessary to determine the true roles of the +869C allele and other polymorphisms in this gene.
Renin–angiotensin–aldosterone system
The local renin–angiotensin–aldosterone system in the lung might play a role in lung fibrogenesis. Angiotensin-converting enzyme (ACE) increases angiotensin II levels, and signalling via angiotensin II receptor type 1 (AT1) has been shown to promote lung fibrosis in rats, possibly through upregulation of TGF-β1 64. The ACE inhibitor captopril has an inhibitory effect on human lung fibroblasts in vitro 65, and inhibition of AT1 has been shown to reduce bleomycin-induced lung fibrosis in rats 64. However, a recent retrospective study from the Mayo Clinic could not demonstrate a beneficial survival effect of ACE inhibition in IPF 66.
A functional insertion/deletion polymorphism in intron 16 of the ACE gene has been found to be responsible for almost half of the variance in serum ACE levels 67, 68. The deletion allele, correlating with high serum ACE levels, has been shown to associate with systemic sclerosis, a disease showing a high propensity for lung fibrosis 69. In addition, Morrison et al. 40 have detected an increased deletion allele frequency in a small cohort of mixed IIP cases (69 versus 54% in controls), which definitively requires further confirmation.
Extracellular matrix
An essential hallmark of IPF is the exorbitant production of extracellular matrix molecules, including collagen, tenascin and proteoglycans. There is clearly an imbalance between the production and degradation of extracellular matrix. In this context, matrix-degrading enzymes, such as matrix metalloproteinases (MMPs) and their inhibitors (tissue inhibitor of metalloproteinases (TIMPs)), are of interest. Zuo 70 has immunohistochemically demonstrated increased expression of MMP-7 (matrilysin) in IPF lungs, which has been confirmed by others 71. Therefore, the MMP-7 gene might be an important candidate gene for further genetic analysis, although the exact mechanism by which this molecule plays a role remains largely unknown. In general, however, MMPs are thought not only to be involved in the breakdown and remodelling that occurs during injury, but may also cause the release of growth factors and cytokines known to influence growth and differentiation of target cells within the lung 72. In addition, Ramos et al. 73 have suggested increased production of TIMPs in IPF, accounting for the inability to degrade matrix. Promoter polymorphisms, which can influence the production levels of these molecules, are therefore of special interest in this respect.
Apoptosis
In normal wound healing, the number of fibroblasts gradually declines as the healing process or active fibrosis is successfully completed or terminated, but seems to persist in lung tissue from patients with IPF 74. An important mechanism herein appears to be apoptosis. In the presence of TGF-β, fibroblasts are protected against apoptosis, but some studies have suggested increased rates of apoptosis 75. In addition, the pattern of expression of pro-apoptotic proteins and inhibitors of apoptosis needs further study before candidate genes can be designated.
In addition to concepts of apoptosis, studies have investigated variations in mechanisms of wound repair, including p53. The p53 gene encodes a nuclear protein that binds to and modulates the expression of genes that play an important role in DNA repair, cell division and cell death by apoptosis. Hojo et al. 76 have demonstrated a close relationship between IPF and the presence of heterogeneous point mutations in fibrotic lung tissue. The occurrence of somatic p53 mutations in IPF is primarily interpreted in the context of the increased tumourigenesis seen in this disease. However, it is also tempting to speculate on a role for somatic mutations in this and other apoptosis-regulating genes in the pathogenesis of IPF itself.
Angiogenic/angiostatic balance
Angiogenesis is the formation of new vessels from pre-existing vasculature. The activation and resolution of angiogenesis is fundamental to wound healing. A microenvironment that encourages angiogenesis has been thought to be a key process in IPF. Although several studies have reported increased angiogenic activity in IPF 77, 78, recent evaluations of interstitial capillary density in IPF lung biopsy specimens could not demonstrate excessive neovascularisation in characteristic fibrotic lesions. However, one of these studies showed increased vascular density in areas of minimal fibrosis 78, 79.
Interestingly, Cosgrove et al. 80 have just described the presence of a potent angiostatic mediator, pigment epithelium-derived factor (PEDF), in IPF lungs. They demonstrated that vascular density is regionally decreased in IPF within the fibroblastic foci, and that, within these areas, PEDF levels were increased, whereas vascular endothelial growth factor levels were decreased. Furthermore, PEDF co-localised with TGF-β1, suggesting that this fibrogenic cytokine might regulate PEDF expression. Taken together, these results suggest a role for dysregulated angiogenesis in the pathogenesis of IPF, and, more specifically, highlight the regional nature of a varied angiogenic response.
PULMONARY FIBROSIS IN SYSTEMIC SCLEROSIS
Systemic sclerosis (scleroderma) is a disease of unknown origin, characterised by excessive deposition of collagen and other connective tissue macromolecules in skin and multiple internal organs, prominent and often severe alternations in the microvasculature, and humoral and cellular immunological abnormalities. One of the major organ manifestations of this clinically heterogeneous disease is pulmonary fibrosis. Scleroderma lung fibrosis appears to be quite distinct from IPF, despite some superficial similarities of clinical, physiological and radiological features. Specifically, response to therapy and survival from this type of diffuse lung disease is better than in IPF even when matched for a variety of indices, including severity of disease at presentation and duration of disease. The histological pattern of scleroderma lung disease is most commonly NSIP 81 by comparison with the UIP pattern of disease that defines IPF. One of the key histological features of NSIP is a homogeneous distribution pattern of combinations of interstitial chronic inflammation and fibrosis. Areas of dense fibrosis and fibroblastic foci, as seen in UIP, are inconspicuous or absent.
The pathogenesis of systemic sclerosis is extremely complex 82, and, unlike in UIP/IPF, immunological inflammation seems to be a crucial component in the pathogenesis. Besides fundamental abnormalities in cells of the immune system, particularly T- and B-cells, alterations in fibroblasts and endothelial cells are intimately involved in the development of the clinical and pathological manifestations of the disease. These abnormalities result in the characteristic triad of pathological changes in systemic sclerosis: 1) humoral and cellular abnormalities, which include the production of numerous autoantibodies, chronic mononuclear cell infiltration of affected tissues, and dysregulation of lymphokine and growth factor production; 2) severe and often progressive cutaneous and visceral fibrosis; and 3) obliteration of the lumen of small arteries and arterioles. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis.
Remarkably, >90% of patients with systemic sclerosis harbour antinuclear antibodies in their serum. There are three predominant antibodies: anticentromere antibodies, antitopoisomerase antibodies (ATAs), and anti-RNA polymerase antibodies. These autoantibodies are almost totally mutually exclusive and define different clinical subsets of the disease with reasonable accuracy 83, 84. In particular, the presence of ATAs (also known as anti-Scl-70 antibodies) in a scleroderma patient is very strongly associated with the risk of development of pulmonary fibrosis (relative risk 17) 83, 84. In addition, ATA positivity was strongly associated with carriage of HLA-DRB1*11 alleles (previously included as part of HLA-DR5), i.e. 39 of 54 (72%) patients who tested positive for ATA carried these alleles versus ∼18% carriership in controls (p = 0.00006) 84. Within HLA-DRB1*11, the allelic subtype *1104 seems to be associated with ATA positivity and the presence of lung fibrosis 85. Gilchrist et al. 84 also showed that HLA DPB1*1301 was tightly associated with ATA presence.
As the early phases of pathogenesis of systemic sclerosis are thought to involve a T-cell-mediated response to an antigenic trigger, possibly epitopes of DNA topoisomerase I, resulting in the production of antibodies directed against this enzyme (ATA), the described HLA associations focus on the major histocompatibility complex (MHC) region on chromosome 6 for the identification of predisposing genetic factors for this disease. The contribution of genetic factors is further supported by the observation of familial clustering of the disease, the high frequency of autoimmune disorders and autoantibodies in family members of patients with systemic sclerosis, and differences in prevalence and clinical manifestations among different ethnic groups 82, 86, 87. Besides MHC-based genes (i.e. HLA genes and pro-inflammatory genes, such as TNF), non-MHC based genes encoding pro-/anti-inflammatory cytokines and chemokines, and genes involved in fibroblast and endothelial cell functioning are also important candidates for a role in the genetics underlying systemic sclerosis. The genes and gene polymorphisms that have been evaluated to date in diffuse systemic sclerosis, i.e. systemic sclerosis associated with pulmonary fibrosis, are discussed below and summarised in table 2⇓.
Genes and gene polymorphisms that have been evaluated in systemic sclerosis (SSc)-associated pulmonary fibrosis
Major histocompatibility complex
Strong associations have been found between HLA-DRB1*11, and also HLA-DPB1*1301, and diffuse systemic sclerosis, and there is some evidence to suggest that it is an amino-acid motif, shared by the different class II susceptibility alleles, that may be pivotal in predisposing to autoantibody formation 84, 85.
However, the MHC region contains multiple extended haplotypes, and, therefore, HLA associations might also point to another nearby gene on chromosome 6p21 that is causally involved in systemic sclerosis. The known linkage disequilibrium between the TNF locus and HLA class II genes has led to studies to define complex haplotypes in that region. Sato et al. 88 have fine mapped across the TNF locus in scleroderma subsets and found an association between the potentially functional TNF -857C>T polymorphism and lung fibrosis. Intergenic haplotype construction revealed, however, that this was fully explained by linkage disequilibrium with HLA-DRB*11. Remarkably, they found that another TNF allele, TNF -863A, was very strongly associated with positivity for anticentromere antibodies and therefore “protective” against pulmonary fibrosis 88. This polymorphism has proven functionality, i.e. the TNF -863A allele is associated with high TNF-α production in vitro 94. This would suggest a differential pathogenetic role for TNF-α across different scleroderma subsets. TNF-related associations might have immense practical implications now that anti-TNF strategies (e.g. infliximab or etanercept) have been proven to be highly effective in rheumatoid arthritis and Crohn's disease, and are increasingly being applied to other chronic inflammatory processes. Therefore, potential intervention with specific strategies, such as TNF-α blockade in systemic sclerosis, might be possible, provided it can be directed to those clinical phenotypes most affected by the effects of this pro-inflammatory cytokine (conceivably anticentromere antibody-positive subjects according to the study of Sato et al. 88).
Non-major histocompatibility complex
Chemokine-related genes
Renzoni et al. 34 studied variations in the gene encoding IL-8 and its receptors (CXCR1 and CXCR2). An increase in the frequency of the CXCR2 +785C and +1208T allele was found compared with controls in the systemic sclerosis patients. However, this was not found to be specific to those with lung fibrosis, being found in systemic sclerosis patients both with and without diffuse lung disease.
Cytokine genes
TGF-β1 is a potent profibrotic cytokine secreted from activated macrophages and T-lymphocytes that may play a central role in the pathogenesis of tissue fibrosis in systemic sclerosis 82. Two studies in systemic sclerosis patients have shown an association between a single nucleotide polymorphism at codon 10 of the gene encoding TGF-β1 and pulmonary fibrosis (notably also associated with IPF progression), which might suggest that a higher production phenotype of this cytokine is particularly related to this clinical phenotype 89, 90.
Extracellular matrix
Variations in genes ontologically related to extracellular matrix homeostasis are of great interest in fibrosing lung diseases as a whole and, especially, lung fibrosis in systemic sclerosis. To date, only weak associations have been reported in the genes encoding fibronectin 1 and secreted protein acidic and rich in cysteine/osteonectin (table 2⇑) 91, 92. A genetically thorough study on the role of the fibrillin 1 gene (FBN1) in systemic sclerosis has been performed by Tan et al. 93. Fibrillin 1 is the major constituent of extracellular microfibrils and has widespread distribution in both elastic and nonelastic connective tissue throughout the body. Mutations in FBN1 are the major cause of Marfan syndrome. In the study of Tan et al. 93, all 69 known exons of FBN1 were sequenced. Five single-nucleotide polymorphisms were identified and tested in Choctaw and Japanese systemic sclerosis patients and controls. On the basis of the genotype results, haplotypes were inferred, and two haplotypes (Hap-5 and Hap-6) were associated with systemic sclerosis in both populations 93. Interestingly, one of these haplotypes (Hap-5) was specifically associated with lung fibrosis. These results are consistent with the hypothesis that FBN1 or a nearby gene on chromosome 15q is involved in systemic sclerosis (lung fibrosis) susceptibility.
CONCLUSION
It is disappointing that stronger associations between genetic polymorphisms and idiopathic fibrosing lung diseases have not yet been found, but the threshold of an exciting era awaits. Better definitions will improve phenotyping and the quality of associations. The large USA familial study will also provide novel candidates, and whether familial disease shows similar predispositions to sporadic disease will be awaited with great interest. The dissection of key associations will undoubtedly require collaboration of the highest quality. Too often, small studies that cannot be reproduced are published. Working together will limit these spurious publications, save time and money and permit true identification of the key issues. The European Respiratory Society is encouraged to continue to facilitate such collaborations both within its organisation and internationally.
- Received November 22, 2004.
- Accepted January 3, 2005.
- © ERS Journals Ltd
References









