





Case history
A 5 month old south Asian male presented to his general practitioner (GP) with a history of recurrent wheeze and noisy breathing of 2 months duration. He had been born at term and his growth and development were normal. There were no significant medical problems in the family, apart from his grandmother's hypothyroidism. He was diagnosed by his GP to have viral wheeze and was started on inhaled salbutamol via a metered dose inhaler plus spacer with no apparent benefit. At this stage, a chest radiograph did not show any abnormality. At 6 months old, the patient was admitted to the local hospital with a history of cough, respiratory distress and stridor, which improved after receiving oral dexamethasone for 2 days.
At 7 months old, he presented at the current authors' hospital with a 2‐day history of cough, wheeze and breathlessness. On examination, the patient was afebrile with moderate respiratory distress and had widespread crepitations and rhonchi. Otherwise, clinical examination was normal. A clinical diagnosis of acute bronchiolitis was made. He needed supplemental oxygen to maintain normal oxygen saturations and was also given a trial of nebulised salbutamol with no apparent benefit.
He was noted to have intermittent noisy breathing, although no obvious upper airway obstructive pathology could be identified. He was making a very slow, but gradual, recovery; however, on day 8, there was clinical deterioration with worsening cough, breathlessness, and noisy breathing. He was found to be tachypnoeic with subcostal and intercostal recession, but his chest was clinically clear on auscultation.
The chest radiograph (fig. 1⇓) showed significant radiological changes compared to the chest radiograph taken at 5 months old.

Chest radiograph of the patient.
The patient was extensively investigated. Table 1⇓ shows a summary of the investigations performed.
Summary of investigations
Biopsies were performed by an open technique. All the samples showed characteristic histological findings (figs 3 and 4⇓⇓), which were later confirmed by immunocytochemistry (fig. 5⇓).
Interpretation
The chest radiograph (fig. 1⇑) shows widened mediastinum. The computed tomography (CT) scan of the chest (fig. 2⇓) at the level of the carina shows a large lobulated heterogeneous soft tissue mass in the anterior mediastinum with some stromal enhancement. The trachea is compressed. There is loss of visualisation of the thymus.

Computed tomography scan of the thorax at carinal level.
The biopsy of the mediastinal mass (fig. 3⇓) shows the characteristic Langerhans cell, which is a large pale cell with convoluted nuclear membrane, indented nuclei and abundant pale cytoplasm. In the same biopsy sample, two eosinophils are also visible.

Biopsy of the mediastinal mass.
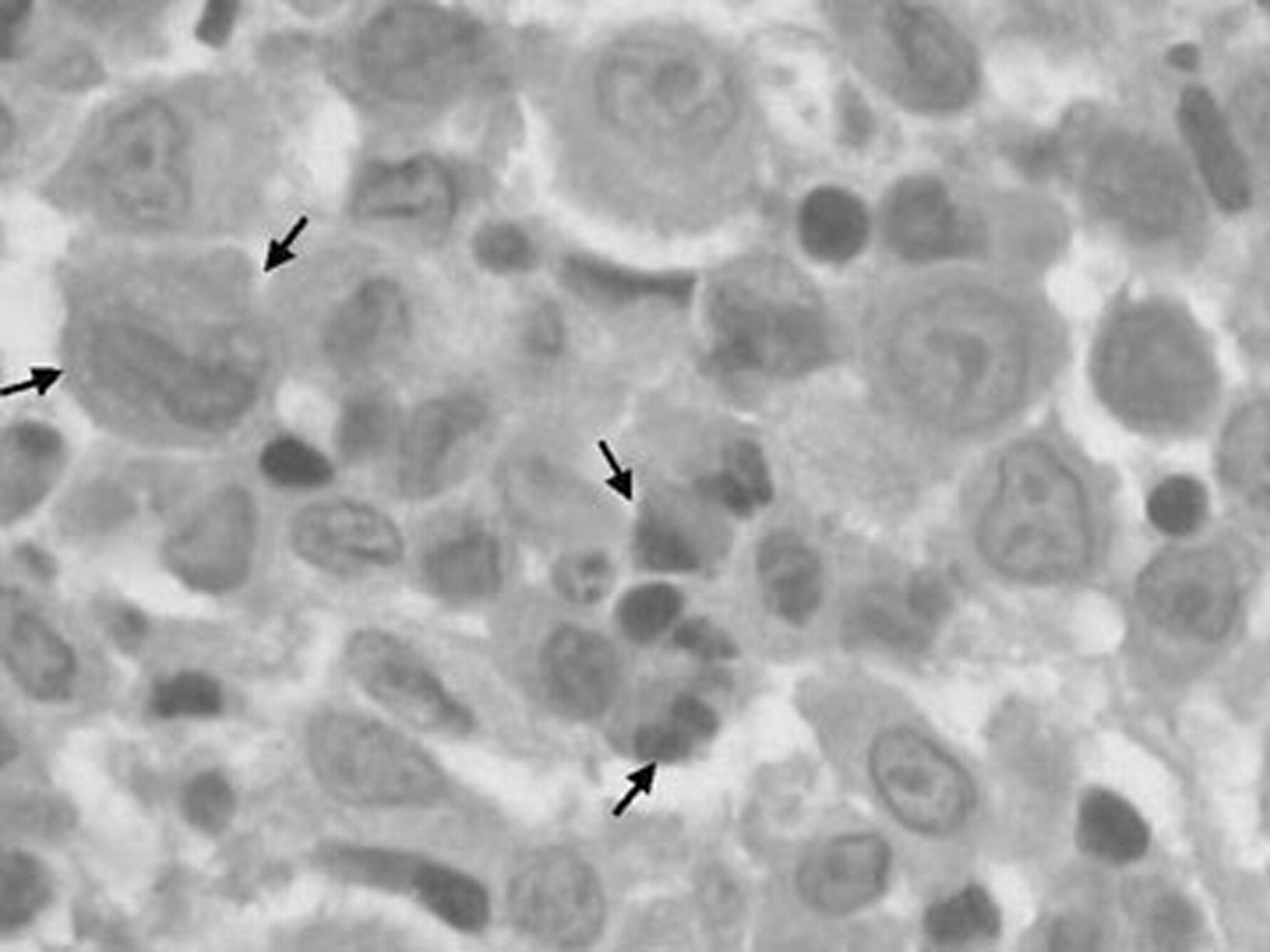
Electron microscopy of the biopsy of the mediastinal mass (fig. 4⇓) shows intracytoplasmic “tennis racket” and rod-shaped Birbeck's granules (arrows). Similar changes were found in the lung biopsy (not shown).

Electron microscopy of the mediastinal mass.
Immunocytochemistry of the biopsy sample (fig. 5⇓) shows characteristic staining pattern with S100 (calcium binding protein soluble in 100% ammonium hydroxide) and CD1A (cluster of differentiation 1a).

Immunocytochemistry of the biopsy sample from the mediastinal mass stained with S100 (a) and CD1A (b).
The radiograph of the left radius (fig. 6⇓) shows osteolytic lesion of the diaphysis and periosteal reaction (arrow).

Radiograph of the left radius.
Diagnosis: Langerhans cell histiocytosis (LCH)
Treatment and clinical course
The patient was started on oral prednisolone and weekly i.v. vinblastine as per LCH treatment protocol. After 10 weeks, a follow-up CT scan of the chest showed significant reduction in the size of the mediastinal mass with minimal residual lesion. The noisy breathing and other chest symptoms improved significantly with the chemotherapy and the bronchodilators were withdrawn. The patient has finished one cycle of chemotherapy and has remained well for 10 months after the diagnosis.
Discussion
The various manifestations of this disease were first recognised as having a common link by Lichtenstein 1 in 1953 and given the name “histiocytosis X”. In 1973, Nezelof et al. 2 firmly established the link between the Langerhans cell and the clinical features.
In 1987, the Writing Group of the Histiocytic Society defined LCH as “an accumulation or proliferation of a clonal population of cells, bearing the phenotype of a Langerhans cell that has been arrested at an early stage of activity and is functionally deficient” 3. The Langerhans cell is a nonpigmented epidermal dendritic cell, which is derived from a multipotent bone marrow stem cell (CD34+), and is a potent antigen-presenting cell. After antigen encounter, the cells migrate to regional lymph nodes where they present the antigen to the paracortical T‐cells 4–7. The most specific markers of Langerhans cells are Birbeck's granules (intracytoplasmic rod- or tennis racket shaped inclusion bodies) and expression of CD1A glycoprotein 5, 8.
The aetiology and pathogenesis have remained an enigma. Proposed theories include environmental, infective, immunological, genetic and neoplastic. Epidemiological studies have shown LCH patients to be consistently under-immunised, suggesting an infective aetiology of LCH 9. The current authors' patient was fully immunised. Thyroid diseases appear to be more common in affected children, as well as other family members 9. In this case, the patient's grandmother had hypothyroidism.
In children, LCH can present from the newborn period to 15 yrs, with a peak incidence at 1–4 yrs, and males are more commonly affected than females 10. Epidemiological data is sparse and only one national incidence estimate of 5.4 per million children has been reported from Denmark 11. Based on this, ∼50–100 new cases in the UK every year can be expected.
There is a wide spectrum of disease activity, ranging from single osteolytic lesion to rapidly fatal leukaemia-like illness. Commonly involved systems are skin, bone, lymph nodes, central nervous system (including diabetes insipidus), ears, gums and lungs. The extent of disease is staged as follows: 1) single system disease; 2) multisystem disease; 3) multisystem disease with organ dysfunction (e.g. abnormal blood film or liver function tests or lung function tests).
Lung disease is frequent in multisystem disease, although the overall prevalence in LCH is estimated at <5%. It may cause respiratory distress with tachypnoea, chest retraction and persistent cough. Primary lung involvement is usually found in adult smokers and, therefore, lung disease is most common in adult LCH patients 5, 12.
Although primary lung involvement is rare in children, its clinical manifestations are similar to those found in adults 5. Children with uncontrolled LCH may develop chronic respiratory failure, presenting with cysts and bullae on radiograph 12.
According to one study, mediastinal mass as a manifestation of LCH is relatively rare (one out of 42 children), whereas pulmonary involvement without mediastinal involvement is more frequent (eight out of 42 children) 13. In the current authors' case, the CT scan did not show lung parenchymal abnormality, but the lung biopsy showed sheets of large pale cells with ovoid or slightly indented nuclei, generally indistinct nucleoli and abundant eosinophilic cytoplasm. These findings are quite characteristic of LCH. There were also a significant number of eosinophils present in the biopsy samples, which supports the diagnosis of LCH. A high resolution CT scan could pick up early pulmonary involvement 13. Definitive diagnosis must include clinical features, characteristic histology and immunocytochemistry (positive for CD1A stain). Calcification within the mass, which was reported in other cases, was absent in the current authors' patient.
Restricted disease carries a good prognosis as the clinical course is usually benign and spontaneous remissions are common. In extensive disease, especially in those with organ dysfunction, the prognosis is guarded with a mortality of 10–15% 10. A normal chest radiograph 2 months before the initial presentation in the current authors' patient points towards the rapid progression of the mediastinal lesion.
Treatment depends on the extent of the disease, age at diagnosis and the presence of organ dysfunction. Steroids can be used for single system disease. In the current authors' case, the patient presented with stridor 1 month before the diagnosis was made and he was treated with oral steroids for 2 days for suspected croup. The stridor could have been due to a pressure effect by the mediastinal mass and steroids could have suppressed the disease progression temporarily. The use of chemotherapeutic agents is reserved for more severe forms 4, 14. There are reports of cavitation developing within the mass while undergoing treatment 15. A wide range of therapies, including bone marrow allografting and stem cell transplantation, have been suggested, but the efficacy of these therapies has not been well documented 16.
This case highlights the need for high index of suspicion for this condition in children presenting with mediastinal mass and respiratory symptoms. Rapid progression of the disease, as in the current authors' case, highlights the need for early diagnosis, as the morbidity and prognosis is highly dependent on the number of organs involved and the extent of organ involvement.
Acknowledgments
The authors are grateful to S. Muller, F. Dickinson and M. Silverman for their help in preparing this report.
- © ERS Journals Ltd










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}