





Abstract
Respiratory failure occurs due mainly either to lung failure resulting in hypoxaemia or pump failure resulting in alveolar hypoventilation and hypercapnia. Hypercapnic respiratory failure may be the result of mechanical defects, central nervous system depression, imbalance of energy demands and supplies and/or adaptation of central controllers.
Hypercapnic respiratory failure may occur either acutely, insidiously or acutely upon chronic carbon dioxide retention. In all these conditions, pathophysiologically, the common denominator is reduced alveolar ventilation for a given carbon dioxide production.
Acute hypercapnic respiratory failure is usually caused by defects in the central nervous system, impairment of neuromuscular transmission, mechanical defect of the ribcage and fatigue of the respiratory muscles.
The pathophysiological mechanisms responsible for chronic carbon dioxide retention are not yet clear. The most attractive hypothesis for this disorder is the theory of “natural wisdom”. Patients facing a load have two options, either to push hard in order to maintain normal arterial carbon dioxide and oxygen tensions at the cost of eventually becoming fatigued and exhausted or to breathe at a lower minute ventilation, avoiding dyspnoea, fatigue and exhaustion but at the expense of reduced alveolar ventilation. Based on most recent work, the favoured hypothesis is that a threshold inspiratory load may exist, which, when exceeded, results in injury to the muscles and, consequently, an adaptive response is elicited to prevent and/or reduce this damage. This consists of cytokine production, which, in turn, modulates the respiratory controllers, either directly through the blood or probably the small afferents or via the hypothalamic-pituitary-adrenal axis. Modulation of the pattern of breathing, however, ultimately results in alveolar hypoventilation and carbon dioxide retention.
This study was supported by the Thorax Foundation, Athens, Greece.
Respiratory failure is a condition in which the respiratory system fails in one or both of its gas exchange functions, i.e. oxygenation of and/or elimination of carbon dioxide from mixed venous blood. It is conventionally defined by an arterial oxygen tension (Pa,O2) of <8.0 kPa (60 mmHg), an arterial carbon dioxide tension (Pa,CO2) of >6.0 kPa (45 mmHg) or both. Therefore, the diagnosis of respiratory failure is a laboratory one, but the important point to emphasise is that these cut-off values are not rigid; they simply serve as a general guide in combination with the history and clinical assessment of the patient.
The respiratory system can be said to consist of two parts: the lung, i.e. the gas-exchanging organ, and the pump that ventilates the lungs 1. The pump consists of the chest wall, including the respiratory muscles, the respiratory controllers in the central nervous system (CNS) and the pathways that connect the central controllers with the respiratory muscles (spinal and peripheral nerves). Failure of each part of the system leads to a distinct entity (fig. 1⇓). In general, failure of the lung caused by a variety of lung diseases (e.g. pneumonia, emphysema and interstitial lung disease) leads to hypoxaemia with normocapnia or hypocapnia (hypoxaemic or type I respiratory failure). Failure of the pump (e.g. drug overdose) results in alveolar hypoventilation and hypercapnia (hypercapnic or type II respiratory failure). Although there is coexistent hypoxaemia, the hallmark of ventilatory failure is the increase in Pa,CO2. Undoubtedly, both types of respiratory failure may coexist in the same patient, as, for example, in patients with chronic obstructive pulmonary disease (COPD) and carbon dioxide retention, or in those with severe pulmonary oedema or asthmatic crisis, who first develop hypoxaemia and, as the disease persists or progresses, hypercapnia appears.

Types of respiratory failure. The respiratory system can be considered as consisting of two parts: 1) the lung; and 2) the pump.
The various types of respiratory failure are presented in a gas tension diagram (fig. 2⇓), which illustrates the various pathways. The solid line represents a respiratory exchange ratio of 0.8. The parallel dotted line shows the corresponding Pa,O2 and Pa,CO2 with an alveolar to arterial oxygen difference (DA‐aO2) of ∼0.67 kPa (5 mmHg), as occurs in normal lung. When a normal subject hyperventilates, alveolar oxygen (PA,O2) and carbon dioxide (PA,CO2) tension and Pa,O2 and Pa,CO2 move down the slope in the direction indicated by the letter H, with rises in PA,O2 and Pa,O2 and falls in PA,CO2 and Pa,CO2. When hypoventilation occurs, in the normal subject, due to drug overdose for example, PA,O2 and Pa,O2 and PA,CO2 and Pa,CO2 move up the slope in the direction shown by the letter D, with falls in PA,O2 and Pa,O2 and rises in PA,CO2 and Pa,CO2. It can be seen that, in the normal lung, when hypercapnia occurs (as in the case of alveolar hypoventilation due to CNS depression), Pa,O2 cannot fall to very low levels. For example, when PA,CO2 increases from 5.3 (40 mmHg) to ∼10.6 kPa (80 mmHg), PA,O2 decreases from ∼13.3 (100 mmHg) to ∼8.0 kPa (60 mmHg). Assuming a DA‐aO2 of 0.67–1.3 kPa (5–10 mmHg), the Pa,O2 is ∼5.3–6.7 kPa (40–50 mmHg). Thus, when the lung is normal, a severe degree of alveolar hypoventilation, resulting in marked carbon dioxide retention, is not associated with excessive hypoxaemia. In lung diseases, however, due to increased DA‐aO2, the same conditions lead to arterial hypoxaemia. Arrow Α, in figure 2⇓, shows a large DA‐aO2 (the horizontal distance between arrow Α and alveolar line D‐H), which is commonly observed in patients with pneumonia, atelectasis or acute respiratory distress syndrome (ARDS). Hyperventilation in these patients leads to very low Pa,CO2. Line Β depicts the pathway of patients with interstitial lung disease or pure emphysema. Line C depicts a mixed state in a patient with lung disease (hypoxaemia) and inadequate alveolar ventilation (V'A). In severe cases (tip of arrow), hypoxaemia is dominant despite hypercapnia and the situation is certainly more dangerous than in pure hypoventilation at an equal Pa,CO2. Patients usually reach line C starting from line Α or B. Patients with COPD or end-stage interstitial lung disease who have remained along the B arrow for a long time move to arrow C as a result of alveolar hypoventilation. Similarly, patients with a gas-exchange abnormality, as shown by arrow Α (acute asthma attack or pulmonary oedema), may move towards arrow B or C as the central controllers or respiratory muscles, or both, become unable to maintain adequate ventilation.

Relation between alveolar oxygen (PO2) and carbon dioxide (PCO2) tension when breathing air (–––––; inspiratory oxygen fraction 21%). The slope of the line depends on the respiratory exchange ratio; in this example, it is assumed to be 0.8. Arterial PO2 (═) is less than alveolar PO2; the difference assumed here is 5 mmHg. A‐C: direction of changes in alveolar gas tensions in various lung diseases. ┘: range of arterial PO2 normally incompatible with prolonged survival (1 mmHg=0.133 kPa).
Pathophysiology of respiratory failure
Hypoxaemic (type I) respiratory failure
Four pathophysiological mechanisms account for the hypoxaemia seen in a wide variety of diseases: 1) ventilation/perfusion inequality, 2) increased shunt, 3) diffusion impairment, and 4) alveolar hypoventilation 2. Ventilation/perfusion mismatching is the most common mechanism and develops when there is decreased ventilation to normally perfused regions or when there are lung regions with a greater reduction in ventilation than in perfusion. With shunt, either intrapulmonary or intracardiac deoxygenated mixed venous blood bypasses ventilated alveoli, resulting in “venous admixture”. Diseases that increase the diffusion pathway for oxygen from the alveolar space to the pulmonary capillaries, decrease capillary surface area or shorten the transit time of the blood through the pulmonary capillaries prevent complete equilibration of alveolar oxygen with pulmonary capillary blood.
In the absence of underlying pulmonary disease, hypoxaemia accompanying hypoventilation is characterised by normal DA‐aO2. In contrast, disorders in which any of the other three mechanisms are operative are characterised by broadening of the alveolar/arterial gradient resulting in severe hypoxemia.
Although changes in V'A can change Pa,CO2 considerably, this is not so for Pa,O2. Increases in V'A modestly increase Pa,O2. Owing to the sigmoidal shape of the oxyhaemoglobin dissociation curve, any effect of increasing ventilation on oxygen saturation is minimal above Pa,O2 of 7.3–8.0 kPa (55–60 mmHg). Hypoxaemia resulting from ventilation/perfusion inequality or diffusion abnormalities can easily be corrected by supplementing inspired oxygen, whereas even very high concentrations of inspired oxygen cannot correct hypoxaemia induced by increased pure shunt.
Hypercapnic (type II) respiratory failure
The respiratory equation
The volume of carbon dioxide eliminated per minute (which in a steady state is equal to that produced by the body (V'CO2)) is dependent on the concentration of carbon dioxide in alveolar gas and οn V'A. This is obvious, since the conducting airways do not exchange gas. Thus V'CO2=V'A×alveolar CO2 concentrationor alveolar CO2 concentration=V'CO2/V'A. Alveolar CO2 concentration is the concentration of CO2 in the alveolar gas. Gas concentration is converted to gas pressure (Pgas) by the equation: Pgas (mmHg)=(%concentration×(barometric pressure-water vapour))/100. At sea level, barometric pressure is 670 and water vapour pressure at 37°C is 47 mmHg. It follows that Pgas (mmHg)=%concentration×713/100.
By using factor k (0.863), the constant of proportionality, the “respiratory equation” is obtained, which relates V'A to Pa,CO2: Since V'A=V'E−V'ds, where V'E is minute ventilation and V'ds dead space ventilation, this relation may be expressed as:
Since V'A=V'E−V'ds, where V'E is minute ventilation and V'ds dead space ventilation, this relation may be expressed as:  where VT is tidal volume and fR respiratory frequency.
where VT is tidal volume and fR respiratory frequency.
Equation 2 states that the Pa,CO2 rises if V'CO2 increases (e.g. hyperthermia) at constant V'A, or when, at a constant V'CO2, V'A decreases by virtue of: 1) a rise in V'ds/VT (by increasing V'ds, decreasing VT or both), 2) a decrease in V'E, and 3) both an increase in V'ds/VT and a decrease in V'E 3, 4.
In daily clinical practice, as a patient becomes hypercapnic, more than one factor generally contributes to the rise in Pa,CO2.
Carbon dioxide production
For a young adult, V'CO2 is ∼200 mL·min−1 (or 110 mL·m2 in males and 96 mL·m2 in females). V'CO2 increases during hyperthermia by ∼14% for each degree Celsius rise in temperature, particularly during muscular activity. During inspiratory resistive breathing, the respiratory muscles, in this respect, may show a V'CO2 of 700–800 mL·min−1 5. In the same manner, shivering or an increase in muscle tone, as occurs in tetanus, leads to excessive V'CO2, with an up to three-fold increase, whereas muscular exercise may increase V'CO2>10‐fold. Under normal conditions, an increase in V'CO2 is detected early by the CNS and is then easily compensated for by increasing V'E to maintain a normal Pa,CO2. However, if a patient's ventilatory capacity is impaired, an increase in V'CO2 greatly stresses the ventilatory system and leads to an increase in Pa,CO2.
Alveolar ventilation
Equations 1 and 2 imply that, at constant V'CO2 and a given V'ds, V'A changes when VT or fR are varied either at constant or reduced total ventilation. This means that there are four possibilities: 1) unchanged total ventilation with decreased fR, 2) unchanged total ventilation with increased fR, 3) decreased total ventilation with decreased fR, or 4) decreased VT.
Under conditions of unchanged total ventilation and decreased fR, for V'E to remain unchanged, VT must increase. This decreases V'ds/VT, thereby increasing V'A and decreasing Pa,CO2.
Under conditions of unchanged total ventilation and increased fR, for V'E to remain unchanged, VT must decrease. Such a change, however, increases the V'ds/VT ratio and, therefore, V'A decreases and Pa,CO2 increases. In the clinical setting, rapid, shallow breathing may well explain carbon dioxide retention in patients with COPD.
Under conditions of decreased total ventilation and decreased fR, the reduction in fR alone, without affecting V'ds/VT, leads to a certain decrease in V'A by virtue of a reduction in V'E.
Under conditions of decreased total ventilation and decreased VT, there is a reduction in V'E caused by reducing the VT (without reducing fR), which results in an increase in V'ds/VT, and, consequently, in a rise in Pa,CO2. Thus, the drop in V'A is expected to be more pronounced than in the aforementioned cases.
Pathophysiology of ventilatory pump failure
There are three major causes of pump failure leading to hypercapnia 6. 1) The output of the respiratory centres controlling the muscles may be inadequate (anaesthesia, drug overdose and diseases of the medulla), resulting in a central respiratory drive that is insufficient for the demand, or the respiratory centres may reflexively modify their output in order to prevent respiratory muscle injury and avoid or postpone fatigue. 2) There may be a mechanical defect in the chest wall, as is the case in flail chest, diseases of the nerves (Guillain-Barré syndrome) and anterior horn cells (poliomyelitis), or diseases of the respiratory muscles (myopathies). Severe hyperinflation, with flat diaphragm and reduced mechanical action of the inspiratory muscles, as in acute asthmatic attack, is one of the most common causes of impaired mechanical performance of the inspiratory muscles. 3) When working under excessive inspiratory load, the inspiratory muscles may become fatigued, i.e. they become unable to continue to generate an adequate pleural pressure despite an appropriate central respiratory drive and an intact chest wall.
It is obvious that, when there is insufficient activation from the CNS, either temporally (e.g. anaesthesia and overdose) or permanently (e.g. diseases of the medulla), respiratory efforts are inadequate and hypoventilation ensues.
Motor output emanating from the CNS needs to be transferred to the respiratory muscles, a process requiring anatomical and functional adequacy of the spinal cord, peripheral nerves and neuromuscular junction. Any disorder along this pathway results in insufficient inflation of the ribcage inadequate to generate subatmospheric pressure, which is essential for the air to flow into the lungs. Mechanical defects of the chest wall (flail chest, kyphoscoliosis and hyperinflation) are entities that predispose to alveolar hypoventilation since they impose additional work on the inspiratory muscles, which have to displace the noncompliant chest wall and lungs.
Since hyperinflation, commonly occurring in diseases characterised by airways obstruction and loss of elastic recoil of the lungs, has multiple adverse effects on inspiratory muscle function, it deserves to be discussed separately.
For humans to breathe spontaneously, the inspiratory muscles must generate sufficient force to overcome the elastic and resistive load of the respiratory system. Furthermore, the inspiratory muscles should be able to sustain the above mentioned load over time and adjust V'E in such a way that there is adequate gas exchange. Fatigue is the inability of the respiratory muscles to continue to generate sufficient pressure to maintain V'A 6. Fatigue should be distinguished from weakness, which is a fixed reduction in force generation not reversible by rest, although muscle weakness may predispose to muscle fatigue.
Fatigue occurs when the energy supply to the respiratory muscles does not meet the demands. Factors predisposing to respiratory muscle fatigue are those that increase inspiratory muscle energy demands and/or decrease energy supplies 7. Energy demands are determined by the work of breathing and the strength and efficiency of the inspiratory muscles (fig. 3⇓)

Respiratory muscle endurance is determined by the balance between energy supplies (S) and demands (D). Normally, the supplies meet the demands and a large reserve exists. Whenever this balance weighs in favour of demands, the respiratory muscles ultimately become fatigued, leading to inability to sustain spontaneous breathing.
The work of breathing increases proportionally with the mean pressure developed by the inspiratory muscles per breath (mean tidal pressure (PI)), expressed as a fraction of maximum inspiratory pressure (PI,max), V'E, duty cycle (inspiratory time (tI)/total respiratory cycle (ttot)) and mean inspiratory flow rate (VT/tI) 6.
PI is increased if the elastic (stiff lungs, pulmonary oedema) or resistive (airways obstruction, asthma) load imposed on the inspiratory muscles is increased. Roussos et al. 8 directly related PI/PI,max with the time that the diaphragm can sustain the load imposed on it (endurance time). The critical value of PI/PI,max that could be generated indefinitely at functional residual capacity (FRC) was ∼0.60. Greater PI/PI,max were inversely related to the endurance time. The critical value of PI/PI,max increases when end-expiratory lung volume increases. Indeed, when lung volume was increased from FRC to FRC plus 50% inspiratory capacity, the critical value of PI/PI,max and the endurance time were diminished to very low values, 25–30% PI,max. Bellemare and Grassino 9 also found that the maximum pressure that can be sustained indefinitely decreases when tI/ttot increases and suggested that the product of PI/PI,max and tI/ttot defined a useful “tension time index” that is related to the endurance time. Whenever the tension time index is below a critical value (0.15 for the diaphragm), the load can be sustained indefinitely.
A weak muscle requires more energy in relation to its maximum energy consumption to perform a given amount of work. The force developed by a skeletal muscle that is sufficient to produce fatigue is a function of the maximum force that the muscle can develop. Any condition that decreases the maximum force decreases the muscle's strength and predisposes to fatigue. Such conditions include atrophy (a probable result of prolonged mechanical ventilation), immaturity, neuromuscular diseases and performance in an inefficient part of the muscle's length/tension characteristics 10, as in a state of acute hyperinflation, during which both the diaphragm and intercostal muscles work at a shorter length.
Finally, muscle efficiency, the ratio of external work performed to energy consumed, is an important factor in energy demands. Inspiratory muscle efficiency is known to fall in patients with hyperinflation. It has been shown that, for the same work of breathing, the oxygen cost is markedly higher in patients with emphysema than in normal subjects 11. This happens, in emphysematous patients, because either some inspiratory muscles may contract isometrically (they consume energy but do not perform work) or the inspiratory muscles are operating in an inefficient part of their force/length relationship: a more forceful contraction is required to produce a given pressure change, and an even greater degree of excitation is required to develop a given force. Thus both conditions lead to increased energy consumption for a given pressure development 12.
Factors determining the inspiratory muscle energy available are muscle blood flow, the Ca,O2 and the blood substrate concentration as well as the ability of the muscles to extract energy (fig. 3⇑).
Diaphragmatic blood flow is essentially determined by the perfusion pressure, which is a function of cardiac output and peripheral vascular resistance, and the vascular resistance of the muscle, which is a function of the intensity and duration of contraction 13. As has been described in animal models, a reduction in cardiac output accompanying cardiogenic or septic shock is a cause of respiratory fatigue leading to severe alveolar hypoventilation, bradypnoea and respiratory arrest 14, 15. Energy supply to inspiratory muscles also depends on the ability of the muscle to increase blood flow in parallel with the increased work. The diaphragm has a greater capacity to increase blood flow than other skeletal muscles 16. However, the amount that the inspiratory muscle blood flow can be increased may be affected by the intensity and duration of muscle contraction. If the respiratory muscles remain contracted throughout the respiratory cycle, as occurs in asthma 17, the overall blood flow to the muscles may be less than that required. In addition, haemoglobin concentration and oxyhaemoglobin saturation influence the aerobic energy supply to the muscle and hence its endurance.
Conditions characterised by inability of the muscles to extract and use energy, such as sepsis or cyanide poisoning, or diminished energy stores and glycogen depletion, as in extreme inanition, may potentially lead to respiratory muscle fatigue.
It is clear from the above discussion that fatigue may occur in a variety of clinical entities that alone or in combination result in an imbalance between respiratory muscle energy supplies and demands. No matter what the causes are, it is well known that fatigue is characterised by loss of force output 18, leading to inability of the respiratory muscles to develop adequate PI during tidal breathing, with consequent decreases in VT and V'E and hypercapnia.
When the respiratory muscles are extensively loaded, however, it is likely that feedback mechanisms modify the central drive, which, by exerting “central wisdom”, alters the ventilatory pattern and serves in reducing the load and alleviating fatigue, thus protecting the ventilatory pump from exhaustion, which, undoubtedly, is a terminal event.
Although there are no data from patients to substantiate the existence of “central wisdom” in ventilatory failure, there is enough evidence to support this notion. The fact that the reduction in VT that followed resistive breathing in animals could be restored promptly to normal by administration of naloxone 19 or bilateral cervical vagotomy 20, as well as the fact that most hypercapnic patients with COPD can achieve normocapnia by voluntarily increasing their ventilation, implies that, although the subjects could increase their ventilation, they chose not to do so.
Indeed, alterations in the pattern of breathing may occur as a result of loading in animals 19, normal subjects 21 and patients during weaning trials 22.
Patients with acute and chronic respiratory failure as well as normal subjects and animals subjected to fatiguing respiratory loads tend to adopt rapid shallow breathing, consisting of a decrease in VT and increased fR, whereas V'E remains constant or increases slightly. Although this pattern may not be efficient in terms of gas exchange, it may reduce the load on the muscle by decreasing the PI developed, thereby preventing fatigue from occurring 23–26. Moreover, in stable patients with COPD and carbon dioxide retention, this pattern of breathing may be sufficient to keep diaphragm contraction below the fatigue threshold 23, 27.
The neurophysiological mechanisms that cause an altered pattern of breathing are not well elucidated. Chemosensitivity-induced alterations in respiratory activity do not appear to be the explanation. Hypoxia- and hypercapnia-induced reductions in expiratory time (tE) are disproportionately greater than reductions in tI, so that tI/ttot increases. Moreover, VT/tI and VT increase rather than decrease 28.
Rapid shallow breathing may be produced by activation of vagal irritant receptors in the airways 29, or may represent a behavioural response to minimise the sense of dyspnoea 30.
Reflexes originating from mechanoreceptors in the contracting ribcage muscles and diaphragm (tendon organs, spindle organs, and type III and IV endings) probably play a role in shaping the rapid shallow pattern of breathing. In deeply anaesthetised animals, stretch of the intercostal muscles or increase in diaphragm tension may abruptly terminate inspiration 31. Activation of endogenous opioid pathways has also been postulated to alter the pattern of breathing, perhaps as a mechanism by which the sense of dyspnoea might be reduced 19, 32–35.
Small-fibre afferents have been widely implicated in the response of central respiratory output to prolonged stresses such as shock, hypoxia, acidosis and vigorous exercise 15, 36, 37. It is possible that, during loaded breathing, afferents, through the small fibres, modulate endogenous opioids as an adaptive response to minimise breathlessness and avoid or delay the onset of respiratory muscle fatigue 38.
Whatever the mechanisms, however, the limit of this strategy is that with rapid shallow breathing V'ds/VT is increased (see The respiratory equation section) with worsening of hypercapnia.
Although failure of the lungs leads mainly to hypoxaemia and failure of the ventilatory pump causes hypercapnia, it has to be emphasised that there are important interactions between these two entities. Furthermore, failure of one part (the lung) is often followed by failure of the second (ventilatory pump). Respiratory diseases that cause hypoxaemia are usually characterised by abnormal lung mechanics, a situation accompanied by increased work of breathing (resistive or elastic) and, therefore, energy demands. Taking into account the fact that the amount of energy available is reduced due to hypoxaemia, it is concluded that lung diseases may result in muscle fatigue and ventilatory failure through imbalance between demands and supplies.
Similarly, patients with diseases that involve the ventilatory pump (myopathies) and who present with hypercapnia are usually characterised by the inability to cough, which leads to accumulation of secretions and possibly atelectasis, a situation eventually aggravating ventilation/perfusion inequality with the result of hypoxaemia.
Pulmonary hyperinflation
In normal subjects at rest, the end-expiratory lung volume (FRC) corresponds to the relaxation volume (Vr) of the respiratory system, i.e. the lung volume at which the elastic recoil pressure of the total respiratory system is zero. Pulmonary hyperinflation is defined as an increase in FRC above the predicted normal value. This may be due to increased Vr as a result of loss of elastic recoil of the lung (e.g. emphysema), or due to dynamic pulmonary hyperinflation, which occurs when FRC exceeds Vr 39.
Whatever its genesis, hyperinflation imposes several constraints on muscle function. For the inspiratory muscles, as for other skeletal muscles, there is an optimum length at which maximum force is developed. This length corresponds closely to FRC or to lower lung volumes 6. With hyperinflation, the respiratory muscle fibres shorten, and thus the force available or the pressure developed for any given level of excitation is decreased. This results in a situation in which, for a given pressure swing necessary for adequate V'A, greater excitation, and thus a greater percentage of the available maximum pressure, is required. This increases the energy consumption for a given workload, and muscle efficiency is thus diminished.
The diaphragm is further compromised by the geometric distortion that hyperinflation induces in the inspiratory muscles. As the diaphragm is flattened and assumes a radius of infinity, Laplace's law (Pdi=2 Tdi/Rdi, where Pdi is transdiaphragmatic pressure, Tdi the tangential tension generated by the diaphragm and Rdi the radius of curvature of the diaphragm) dictates that the diaphragm is no longer able to convert tension to pressure efficiently 40. Apart from this, as the diaphragm flattens, the zone of opposition is reduced. This zone is the region of the costal diaphragm that abuts the lateral chest wall. In normal subjects, when the diaphragm contracts, the increase in abdominal pressure is transmitted to the ribcage through this zone, thereby facilitating thoracic expansion. Therefore, as lung volume increases, the contribution of diaphragmatic contraction to chest wall expansion is minimised.
Furthermore, hyperinflation alters the spatial orientation of diaphragmatic costal and crural fibres, forcing them to be arranged in series and perpendicularly with regard to the chest wall. During inspiration, contraction of these perpendicularly oriented fibres results in paradoxical inward movement of the lower ribcage (Hoover's sign).
As with the diaphragm, the length/tension characteristics and geometry of the thorax place the inspiratory intercostals or accessory muscles at a disadvantage during hyperinflation 6.
The work of breathing, almost invariably increased by the abnormal lung mechanics that lead to hyperinflation, is further increased by hyperinflation itself. Consequently, energy demands are increased, whereas energy supply may be limited due to sustained contraction of the respiratory muscles. The combination of increased work, decreased strength, decreased efficiency and decreased energy supply during hyperinflation puts the respiratory muscles at greater risk and makes them particularly prone to fatigue.
Ventilatory failure in clinical conditions
Hypercapnic respiratory failure may occur either acutely, insidiously or acutely upon chronic carbon dioxide retention.
Hypercapnia of acute onset
The pathological entities resulting in acute carbon dioxide retention are anatomical and functional defects of the CNS, impairment of neuromuscular transmission and mechanical defect of the ribcage, as well as conditions leading to fatigue of the respiratory muscles (table 1⇓). Mechanisms responsible for carbon dioxide retention are both decreasing V'E and increasing V'ds/VT.
Causes of alveolar hypoventilation, acute onset
Depression of the CNS due to pharmacological agents, infections or head trauma leads to hypoventilation due to impaired respiratory drive.
Mechanical defects of the chest wall (flail chest and acute hyperinflation), neuromuscular diseases (bilateral diaphragmatic paralysis, myasthenia Gravis, botulism and Guillain-Barré syndrome) and pharmacological agents such as curare may result in acute hypercapnia.
Acute hypercapnia and eventually fatigue may occur in every clinical condition that is characterised by an increase in the mechanical load of breathing, and, consequently, in the energy demands that cannot be met by the energy supplies. Under these conditions, the CNS may reflexively adjust its outgoing signals in order to avoid complete depletion of vital chemicals within the muscle cell 18 and/or overt fatigue. In this way, VT is reduced by diminishing tI in order for the PI to be diminished and therefore the energy demands per breath (tension time index per breath is reduced). In addition, with small tidal breaths, the respiratory muscles operate at a more optimal length that does not substantially affect their geometry. The reduction in VT is compensated for, at least in the beginning, by increasing the fR so that V'E is maintained or increased. Such a situation leads to rapid and shallow breathing 41, 42, increased V'ds/VT and hypercapnia.
This fR, however, is no longer optimal, and, for the same V'A, the energy demands are increased. Thus, although shortening of tI seems to be a better option in terms of energy demands, if coupled with high fR and possibly inadequate energy supply, as occurs during strenuous contractions, it may lead to muscle fatigue. Pressure generated by the inspiratory muscles then decreases with consequent decreases in VT and V'E and an increase in V'ds/VT 42, with a subsequent reduction in V'A that is accompanied by an increase in Pa,CO2. At a later stage, tI increases again and the fR gradually decreases, resulting in a further drop in V'E 43. In extreme fatigue, the CNS reduces the output signals per breath, leading to respiratory arrest 14.
Thus alveolar hypoventilation with consequent hypercapnia is the result of a reduced tension time index, which may be due to muscle fatigue or possibly adaptation of the CNS before the development of overt fatigue.
In an acute asthmatic attack, characterised by severe airways obstruction, the rapid shallow breathing associated with the increased elastic and resistive inspiratory load increases the work of breathing and consequently the energy demands. Reduced strength and efficiency of the respiratory muscles due to hyperinflation, as well as impaired blood supply to them due to strong muscle contractions, probably leads to decreased PI,max, while, at the same time, PI is increased. Increased PI/PI,max leads to dyspnoea 30, and, potentially, if a critical value is crossed, fatigue. Such a situation forces alveolar hypoventilation by reducing VT, either as a protective mechanism for the muscles, since there is enough evidence that strenuous breathing causes muscle injury 44, 45, or as a consequence of fatigue of the muscles.
In noncardiogenic (ARDS) and cardiogenic pulmonary oedema, energy demands are increased due to increased elastic work and hyperventilation, whereas energy supply is diminished due to hypoxaemia in the former, and hypoxaemia as well as low cardiac output in the latter. In such situations, the respiratory muscles may fail.
Acute hypercapnic respiratory failure may also develop in patients with acute respiratory failure receiving mechanical ventilation during the weaning trial. The pathophysiological pathways that lead to carbon dioxide retention have been better studied in this group of patients, since, in other clinical conditions, it is difficult, if not unethical, to delay therapeutic interventions in order to document the physiological parameters.
In the first relevant and very significant study, performed by Cohen et al. 43, it was noticed that the majority of patients who failed the weaning trial presented with tachypnoea, acidosis and fatigue of the respiratory muscles. The above authors, as well as others subsequently, proposed that fatigue may be the final common pathway leading to hypercapnic respiratory failure during discontinuation from mechanical ventilation 22. Further research regarding this issue has shown that rapid shallow breathing (with the consequent rise in V'ds/VT), as well as a significant increase in the elastic and resistive load of the respiratory muscles, are the major causes of carbon dioxide retention in these patients 22, 24. Although there is some controversy regarding the exact role of inspiratory muscle fatigue during weaning failure, there are enough data offering significant support in favour of fatigue. Goldstone et al. 46, upon measuring maximum relaxation rate during weaning, observed a decline in those patients failing to be weaned, a finding suggesting that a fatigue process is initiated peripherally in the respiratory muscles. Furthermore, using criteria similar to Cohen et al. 43, Brochard et al. 47 found that patients who failed to be weaned exhibited electromyographic signs of fatigue during spontaneous breathing followed by a decrease in VT, increase in fR and development of hypercapnia. Disuse atrophy of the respiratory muscles, a well documented condition after short- and long-term mechanical ventilation 48, malnutrition, the decreased perfusion that is often seen in these patients, and also decreased strength and efficiency of the muscles due to hyperinflation are conditions that decrease respiratory muscle capacity. These, coupled with increased mechanical load, are situations leading to respiratory muscle fatigue.
The fact that the weaning trial was interrupted and ventilatory support resumed prior to inspiratory muscle exhaustion once the patients presented with clinical signs of respiratory distress (in the period of time before the appearance of muscle fatigue) may explain why some investigators failed to document long-lasting diaphragmatic fatigue in patients failing to be weaned from the ventilator 49. Furthermore, it should be emphasised that fatigue is not an “all or nothing phenomenon” 50; rather the impairment in contractility is more likely to exist in the form of a continuum.
In most neuromuscular diseases with acute onset (e.g. diaphragmatic paralysis or the effects of poisons such as organophosphates), weakness of the respiratory muscles is a very common feature, and leads to ventilatory failure. Weakness could also be the result of muscle atrophy, malnutrition, electrolytic disorders or immature respiratory muscles, as in newborn babies. In these situations, the remaining normal muscle cells cannot develop sufficient force to maintain adequate V'A, and they eventually become fatigued.
Insidious onset of hypercapnia
Patients with chronic hypercapnia have to breathe against increased forces imposed by either the lung (as in bronchitis and emphysema) or chest wall (as in kyphoscoliosis, extreme obesity or neuromuscular disorders), or both (as in scleroderma and polymyositis) (table 2⇓).
Causes of alveolar hypoventilation, insidious onset
Although chronic hypercapnia is most frequently seen in COPD, where it is associated with an ominous prognosis 51, the mechanisms leading to its occurrence are not completely understood. The relationship of Pa,CO2 with indices of airways obstruction or ventilation/perfusion mismatch is weak 52, suggesting that factors other than lung pathology may be operative.
More than half a century ago, two hypotheses were proposed in order to explain chronic hypercapnia in these patients. Scott 53, in 1920, suggested that impaired ventilation is the result of chemical factors that influence the respiratory drive through changes in the chemical environment of the respiratory centres. Christie 54, in 1934, proposed that ventilatory insufficiency is primarily the result of abnormal respiratory mechanics, and that the respiratory muscles are unable to perform the necessary work to provide adequate ventilation and hence carbon dioxide levels rise. The fact that most hypercapnic COPD patients can achieve normocapnia by voluntarily increasing their ventilation 55 makes the two hypotheses above untenable. Given the increase in V'ds/VT ratio that occurs in COPD, Pa,CO2 could be maintained at or near normal levels provided that V'E can be preserved at a sufficiently high level. The fact that, in steady-state COPD patients, deep breathing brings the respiratory muscles near to fatigue and cannot be tolerated for more than a few minutes 27 probably indicates that hypercapnic COPD patients choose to act as “wise fighters”, who, instead of increasing their ventilation (an option that could potentially lead to muscle fatigue), choose to hypoventilate.
The problem with maintaining V'E above normal levels is that this is associated with significant mechanical impediments to breathing. Since COPD patients are characterised by increased airflow resistance and reduced dynamic compliance, the resistive and elastic loads are increased and hence the inspiratory muscles have to generate higher forces to inflate the lung. The emphysematous changes in the lung cause hyperinflation, which forces the inspiratory muscles to operate at shorter than normal lengths and reduces their ability to lower the intrathoracic pressure. More importantly, the dynamic hyperinflation that develops in these patients due to limitation of expiratory flow imposes a severe strain on the respiratory muscles because of the additional load that is placed on them (intrinsic positive end-expiratory pressure (PEEPi)) and because of impairment of their operating length and geometry.
Subsequently, the balance between the mechanical impediments to breathing and the capacity of the inspiratory muscles to cope with them, expressed by PI/PI,max, is shifted in an unfavourable direction (more work/less muscle reserve). Under such conditions, COPD patients choose to reduce PI by reducing VT. A reduced VT may reflect reduced central respiratory drive, or, alternatively, mechanical limitation and/or inspiratory muscle dysfunction.
A subnormal respiratory output has long been postulated as a mechanism of hypercapnia in patients with COPD. However, neural drive assessed by mouth occlusion pressure has been found to be higher in COPD patients than in normal subjects 56, although no significant differences were found between normocapnic and hypercapnic patients. In addition, neural drive assessed by surface electromyographic activity of the diaphragm was also found to be increased in both normocapnic and hypercapnic COPD patients 57. Furthermore, the voluntary drive to breathe has been shown not to be decreased in hypercapnic COPD patients 58. Consequently, although, in these patients, the respiratory drive to breathe is increased, they are better off shortening the tI, a process that results in a lower VT.
The reduction in VT is largely offset by an increase in the fR, such that V'E is well preserved. However, rapid shallow breathing has undesirable consequences. The increased fR aggravates dynamic hyperinflation and increases V'ds/VT. The pattern of breathing in COPD patients has been examined in several studies. Comparing normal persons with hypercapnic and normocapnic stable COPD patients, Sorli et al. 56 noted that hypercapnic patients breathed faster and more shallowly than either normal persons or normocapnic subjects and so, at equal V'E, V'ds/VT was higher in the carbon dioxide retainers. Decreased VT and increased V'ds/VT were also found by Begin and Grassino 59 in severely hypercapnic COPD patients. The authors suggested that chronic alveolar hypoventilation is likely to develop in COPD patients who have a combination of high inspiratory loads (resistive and elastic) and greater hyperinflation, factors that effectively reduce PI,max, thus increasing the fraction of force developed/force available for breathing (less efficient muscles). These data were confirmed by Gorini et al. 60, who found that VT was related directly to tI, indicating that a small VT was primarily the consequence of alterations in respiratory timing.
The mechanisms leading to alterations in respiratory timing in patients with COPD have not yet been clearly defined. Changes in the pattern of breathing may represent a behavioral response in order to minimise the sense of dyspnoea. The sense of dyspnoea is a complex perceptual construct, probably multifactorial 30. Studies indicate that the sense of breathlessness increases with increases in the intrathoracic pressure required to maintain airflow and VT, tI (relative to ttot) and fR. Thus, at a given V'E and set of respiratory mechanics, the pattern of breathing determines the intensity of breathlessness.
In the study of Begin and Grassino 59, in the COPD patients who presented with gross hyperinflation (residual volume/total lung capacity 76%) and hypercapnia, PI/PI,max was markedly increased compared to normal subjects (27 versus 10%), a value highly probable to predispose the muscles to fatigue 4. Thus it could be suggested that, as the disease progresses, the critical level of power output for muscle fatigue is exceeded in order to permit the patient to maintain adequate V'A. Thus it seems evident that, when the muscles become unable to develop enough force, the activation system comes into play and may alter the pattern of breathing in an attempt to optimise the performance of the muscles and possibly postpone or prevent severe fatigue.
Although the underling mechanisms are not known, it is speculated that afferents from the small fibres stimulated by the heavy work (ergoreceptors, type III) or noxious substances (nociceptors, type IV) modify CNS output.
Acute-on-chronic respiratory failure
Acute deterioration in a patient with chronic respiratory failure is termed acute-on-chronic respiratory failure. Patients may present with worsening dyspnoea, deteriorating mental status or respiratory arrest after relatively minor, although often multiple, insults. Acute-on-chronic respiratory failure is usually seen in patients known to have severe COPD. Patients with severe but stable COPD exist in a very critical balance between increased demands and limited reserves. Any factor that potentially interferes with this balance (either increase in demands or decrease in reserves) leads to respiratory muscle fatigue and acute respiratory failure.
Patients with COPD experience an increased respiratory system load due to abnormal airway resistance and respiratory system elastance. The increased resistance is caused by bronchospasm, airway inflammation or physical obstruction by mucus and scarring. The most significant contributor to the elastic load is dynamic hyperinflation that develops whenever the tE is insufficient to allow the lungs to deflate to Vr prior to the next inspiration. This tends to occur under conditions in which expiratory flow is impeded (increased airway resistance) or when the tE is shortened (increased fR) 39, 61. Expiratory flow may also be retarded by other mechanisms such as persistent constriction of the respiratory muscles during expiration. Most commonly, however, dynamic pulmonary hyperinflation is observed in COPD patients who exhibit expiratory flow limitation during resting breathing and plays a paramount role in causing respiratory failure.
When breathing takes place at lung volumes greater than Vr, a positive elastic recoil pressure called PEEPi remains at end-expiration.
When PEEPi is present, the inspiratory muscles have to generate greater effort to overcome an equal amount of pressure before airflow starts. In this respect, PEEPi acts as an inspiratory threshold load which increases the static elastic work of breathing. On average, it has been found that the inspiratory work due to PEEPi represents 57% of the overall increase in the work of breathing exhibited by COPD patients relative to normal subjects 62.
Owing to hyperinflation, tidal breathing occurs at a steeper portion of the pressure/volume curve of the lung (increased elastance), increasing the inspiratory load. Apart from increasing elastic loads, dynamic hyperinflation is accompanied by a concomitant decrease in the effectiveness of the inspiratory muscles as pressure generators, because the inspiratory muscle fibres become shorter and their geometric arrangement changes (see Pulmonary hyperinflation section). However, patients presenting with acute-on-chronic respiratory failure may show not only worsening hyperinflation but also other conditions that cause muscle weakness (protein/calorie malnutrition and steroid myopathy). Figure 4⇓ shows a schematic representation of the sequence of responsible mechanisms that lead to acute-on-chronic respiratory failure in COPD patients.

Schematic representation of the sequence of responsible mechanisms that lead to acute-on-chronic respiratory failure in patients with chronic obstructive pulmonary disease. ttot: total respiratory cycle; tI: inspiratory time; te: expiratory time; Raw: airway resistance; EL,dyn: dynamic elastance of the lung; PEEPi: intrinsic positive end-expiratory pressure; ↓: decrease; ↑: increase.
Acute ventilatory failure in these patients is usually triggered by airway infection. The increased fR, which is invariably present in acutely ill COPD patients 39, 63 due to shortened tE, further exacerbates dynamic hyperinflation, which promotes an increase in the static elastic work of breathing (due to both PEEPi and decreased lung compliance). At the same time, an acute increase in airway resistance (bronchoconstriction and copious secretions) causes an increase in the resistive work of breathing. The increased work of breathing associated with this impaired muscle effectiveness leads to an increase in energy requirements, which, at a critical point, exceeds the diminished energy available (hypoxaemia and impaired diaphragmatic blood flow due to forceful contractions) and respiratory muscle fatigue ensues with a further increase in Pa,CO2.
Perspectives: hypothalamic-pituitary-adrenal axis and ventilatory failure
Although the processes leading to acute hypercapnic respiratory failure have been thoroughly elucidated, the pathophysiological mechanisms responsible for chronic carbon dioxide retention are not yet clear. The fact that patients with COPD who most probably could increase their ventilation 55 choose not to and as a result develop chronic hypercapnia permits the speculation that it might be “natural wisdom” that protects these patients from the disastrous consequences of their disease, but with the inevitable cost of hypercapnia.
The study and eventually the partial elucidation of the multiplicity of mechanisms functioning through “natural wisdom” would probably be an interesting and satisfying experience for the investigator in this field.
Since the intense strain of the diaphragm that is usually required for adequate V'A under conditions of increased load and/or reduced efficiency has been proven to lead to structural damage of the respiratory muscles 44, 45, it could be assumed that a protective mechanism might exist in order to protect the respiratory muscles from damage.
Although no indisputable evidence exists to prove the mechanisms involved in chronic carbon dioxide retention, there are sufficient data to permit speculation concerning the way in which peripheral mechanical or chemical stimuli are transferred and modify the pattern of breathing 36, 37.
Until the early 1970s, the respiratory central nervous discharge had been considered to be affected predominantly by central and peripheral chemoreceptors and vagal afferents, and to a lesser extent by muscular afferents, with a role that seemed of minor importance. The effects of sensory information traveling via the phrenic nerves on the central respiratory centres were not very well elucidated, although it was known that nonmyelinated fibres constituted a large proportion of the entire phrenic nerve 64, 65. The functional existence of small myelinated and nonmyelinated phrenic afferent fibres (types III and IV respectively), as well as their influence on central inspiratory neural activity, was confirmed in anaesthetised cats, in which it was shown that selective stimulation of these fibres influences the respiratory rhythm (altering the tI/ttot) and hence central inspiratory activity 36. Under states of severe thoracic stress (muscle tension, local ischaemia and accumulation of toxic metabolites), afferents from the respiratory muscles may play a predominant role in the genesis of the particular breathing pattern. As has been shown, the characteristic response to breathing against a fatiguing load or in a state of reduced respiratory blood flow is tachypnoea, initially followed by bradypnoea and respiratory arrest 14, 37. Since this response is not affected in animals, by either vagotomy, eliminating vagal afferents, or cross-perfusion of the head, eliminating chemoreceptor afferents, it seems reasonable to suggest that afferent information from type III and IV receptors would potentially increase their effect on the central respiratory controllers and alter ventilatory timing 36, 37.
The influence of afferent phrenic fibres on breathing pattern, as well as possible stimuli triggering them, has been studied in anaesthetised animals during eupnoea or fatigue trials 38, 66. In conscious goats, strenuous resistive breathing was associated with a biphasic electromyographic response in the diaphragm 67, consisting of an initial and immediate increase (facilitation) followed by a partial decrease (inhibition). Both responses have been attributed to small afferent fibre activation, initially causing facilitation, and, in a time- and/or intensity-dependent manner, partial inhibition through the elaboration of β‐endorphin. A direct link between stimulation of group III and IV afferents and the central elaboration of endogenous opioids has also been suggested by Kumazawa et al. 68, who showed that, in anaesthetised dogs, respiratory depression persisted even after withdrawal of afferent fibre stimulation, whereas the magnitude of the respiratory depression was significantly reduced by naloxone. Other investigators have confirmed these results in cats and demonstrated that the phenomenon involved a supraspinal mechanism 69 which could be prevented by pretreatment with naloxone 32, thus strongly indicating involvement of endogenous opioid pathways.
The role of exogenous or endogenously generated opioids as neurotransmitters or neuromodulators of a complex inhibitory system of respiration has been extensively studied. When injected into the cisternal cerebrospinal fluid, heroin caused a reduction in VT 33, indicating a direct inhibitory effect of this opioid on respiration. In COPD patients whose respiratory responses to flow resistive loading were absent, the responses could be immediately restored by administration of naloxone 34, suggesting that the chronic increase in airway resistance in these patients generated endogenous opioids as an adaptive response, which served to lessen the stress of prolonged dyspnoea. Acute short-term inspiratory resistive loading resulted in a reduction in VT and mean inspiratory flow rate in unanaesthetised goats, as well as an increase in the levels of cisternal cerebrospinal fluid imunoreactive β‐endorphin 19. Both VT and mean inspiratory flow rate could be transiently increased by naloxone administration, demonstrating that a proportion of these effects could have been meditated by elaboration of endogenous opioids. These data imply that endogenous opioids caused a progressive decline in the discharge rate of the respiratory centres that allow a reduction in inspiratory activity per breath in order to minimise the work of the respiratory muscles, since a reduced VT requires less pressure development, and delay or prevent the onset of overt muscle fatigue.
Although strenuous inspiratory resistive breathing has been found to produce β‐endorphin not only in animals but also in humans 35, 70, the source of these substances remains elusive, and both central sites such as the hypothalamic-pituitary-adrenal axis and peripheral sites such as the spinal cord and peripheral nerves have been implicated 67.
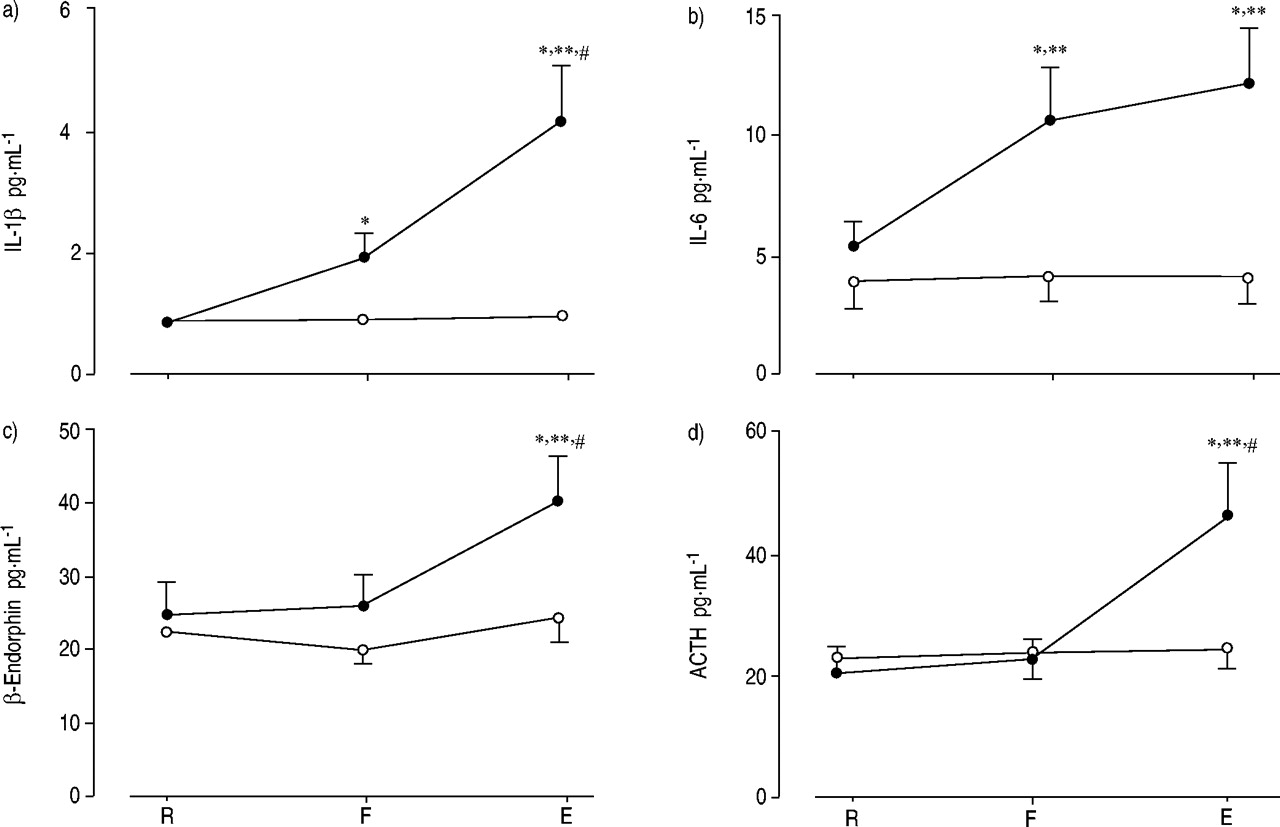
Recently, it was shown that, in normal human volunteers, strenuous resistive breathing leads to a significant rise in the levels of the pro-inflammatory cytokines interleukin (IL)‐1β, IL‐ 6, adrenocorticotropic hormone (ACTH) and β‐endorphin 71. The strong relationships between the rise in β‐endorphin and ACTH levels and the preceding increase in circulating IL‐6 levels allowed the authors to suggest that pro-inflammatory cytokines, and especially IL‐6, are responsible for activation of the hypothalamic-pituitary-adrenal axis, leading eventually to an increase in plasma β‐endorphin and ACTH levels, given that both are derived from post-translational modification of the same molecule, pro-opiomelanocortin 72, and are concomitantly secreted by the pituitary gland 73, 74. It is tempting to suggest that the mechanism accounting for the increase in β‐endorphin and ACTH levels could be the stimulation of small afferent nerve fibres by the cytokines that are produced during resistive breathing (fig. 5⇓). Indeed, global depletion of small afferent fibres inhibits the plasma ACTH response to intravenous IL‐1β 75.

Mean plasma levels of: a) interleukin (IL)‐1β; b) IL‐6; c) β‐endorphin; and d) adrenocorticotropic hormone (ACTH) at rest (R), the point at which subjects could not generate the target maximum inspiratory pressure (F) and the end of resistive breathing (E) (•: high load; ○: moderate load). Data are presented as mean±sem. *: p<0.05 versus R; #: p<0.05 versus F; **: p<0.01 versus moderate load. (Reproduced with permission from 66).
Although the stimuli for the production of the cytokines are not known, reactive oxygen species produced within the respiratory muscles during fatiguing resistive breathing 76, 77 could probably be responsible. It has very recently been discovered that antioxidant administration in healthy volunteers significantly blunts the strenuous resistive breathing-induced cytokine response 78: the tumour necrosis factor‐α response was abolished, IL‐1β became undetectable, and the IL‐6 response was blunted. Thus it is possible that plasma cytokine induction during resistive breathing is differentially regulated by various stimuli, some of them being common (reactive oxygen species), whose relative importance varies with each respective cytokine.
Even though the present authors acknowledge that the time profile (brief) and intensity (much greater) of the increased resistance in the above mentioned models are different from those in patients with chronic carbon dioxide retention, given that strenuous breathing causes diaphragm muscle fibre injury consisting of membrane damage, sarcomere disruption 44, 45 and lactic acid production 38, the following hypothesis could be arrived at (fig. 6⇓). Strenuous resistive breathing, through reactive oxygen species, induces plasma cytokine production, especially IL‐6, which in turn modulates the respiratory controllers either directly through the blood or probably the small afferents or via the hypothalamic-pituitary-adrenal axis. The ensuing stimulation of the hypothalamic-pituitary-adrenal axis by the cytokines might have a dual purpose: the ACTH response may represent an attempt of the organism to reduce the injury occurring in the respiratory muscles through the production of glucocorticoids by the adrenals 79. At the same time, production of β‐endorphin decreases the activation of the respiratory muscles 35 and alters the pattern of breathing 19 in an attempt to reduce and/or prevent further injury at the cost of V'A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Possible loop between respiratory muscles and central controllers: reactive oxygen species (ROS), interleukin (IL)‐6, small fibres, hypothalamic-pituitary-adrenal axis, adrenocorticotropic hormone (ACTH) and endorphins. POMC: pro-opiomelanocortin.
Conclusions
The respiratory system consists essentially of two parts: a gas exchanging organ, the lungs, and a pump that ventilates the lungs. The pump consists of the chest wall, the respiratory muscles that displace the chest wall, the respiratory centres of the CNS that control the muscles and the nerves connecting the centres to the muscles. Both parts of the system are vital. In general, failure of the gas exchanging function due to lung disease (i.e. ARDS, pneumonia and pulmonary emboli) results mainly in hypoxaemia with normocapnia or hypocapnia (type I respiratory failure). Failure of the pump (i.e. CNS depression, weakness and trauma), which also causes hypoxaemia, leads to hypoventilation and hypercapnia which is the hallmark of ventilatory failure (type II respiratory failure).
Hypercapnic respiratory failure may occur either acutely, insidiously or acutely upon chronic carbon dioxide retention. In all these conditions, pathophysiologically, the common denominator is reduced V'A for a given V'CO2.
Mechanical disorders are often a cause of alveolar hypoventilation. Disorders with hypoventilation can easily be seen in chest wall trauma (flail chest), excessive hyperinflation (in hyperinflated patients with obstructive lung disease) and kyphoscoliosis. Neuromuscular disorders in which the patients present with a weakness, as, for example, in overdose, CNS lesions and neuromuscular diseases, lead to alveolar hypoventilation either acutely or chronically. Imbalance of energy demand and supply may eventually lead to respiratory muscle fatigue (i.e. shock), and, in turn, to reduced ventilation resulting in hypercapnia.
Extensive discussion over many years has not provided an answer as to why some patients develop chronic hypercapnia as in chronic obstructive pulmonary disease, kyphoscoliosis and neuromyopathies. The most attractive hypothesis in this disorder is the theory of “natural wisdom”. Patients facing a load have two options: either to push hard in order to maintain normal arterial oxygen and carbon dioxide tensions at the cost of eventually becoming fatigued and exhausted or breathe at a lower minute ventilation, avoiding dyspnoea, fatigue and exhaustion but at the expense of reduced alveolar ventilation. Credence is lent to this latter option, which the present authors favour, by most recent work. In brief, it is possible that a threshold inspiratory load may exist, which, whenever exceeded, results in injury to the muscles, and, consequently, an adaptive cytokine and hormonal response is elicited to prevent and/or reduce the damage. This, however, culminates in alveolar hypoventilation and carbon dioxide retention.
- Received April 7, 2003.
- Accepted August 7, 2003.
- © ERS Journals Ltd
References









