





Abstract
The aim of this study was to investigate if mannitol inhalation, as a model of exercise-induced bronchoconstriction (EIB), causes mast cell activation and release of mediators of bronchoconstriction.
Urinary excretion of previously identified mediators of EIB was investigated in association with mannitol-induced bronchoconstriction. Twelve asthmatic and nine nonasthmatic subjects inhaled mannitol and urine was collected 60 min before andfor90 min after challenge. The urinary concentrations of leukotriene (LT)E4, the prostaglandin (PG)D2 metabolite and the mast cell marker 9α,11β‐PGF2 weremeasured by enzyme immunoassay. Nτ‐methylhistamine was measured by radioimmunoassay.
In asthmatic subjects, inhalation of a mean±sem dose of 272±56 mg mannitol induced a reduction in forced expiratory volume in one second (FEV1) of 34.5±2.1%. This was associated with increases in urinary 9α,11β‐PGF2 (91.9±8.2 versus 66.9±6.6 ng·mmol creatinine−1, peak versus baseline) and LTE4 (51.3±7.5 versus 32.9±4.7). In nonasthmatic subjects, the reduction in FEV1 was 1.0±0.5% after inhaling 635 mg of mannitol. Although smaller than in the asthmatics, significant increases of urinary 9α,11β‐PGF2 (68.4±6.9 versus 56.0±5.8 ng·mmol creatinine−1) and LTE4 (58.5±5.3 versus 43.0±3.3 ng·mmol creatinine−1) were observed in the nonasthmatic subjects. There was also a small increase in urinary excretion of Nτ‐methylhistamine in the nonasthmatics, but not in the asthmatics.
The increased urinary levels of 9α,11β‐prostaglandin F2 support mast cell activation with release of mediators following inhalation of mannitol. Increased bronchial responsiveness to the released mediators could explain the exclusive bronchoconstriction in asthmatic subjects.
- bronchial provocation
- leukotriene E4
- mannitol
- Nτ‐methylhistamine
- 9α
- 11β‐prostaglandin F2
- urinary mediators
This study was supported by the National Health and Medical Research Council of Australia (J.D. Brannan), the Swedish Medical Research Council (12754, 13047, 9072), the Swedish Society for Health Care and Allergy Research (Vårdalstiftelsen), the Swedish Heart Lung Foundation, the Swedish Asthma and Allergy Research Foundation and Karolinska Institutet.
The mechanism whereby exercise causes the airways to narrow is by the osmotic and thermal consequences of evaporative water loss in humidifying the inspired air 1. Release of mediators from inflammatory cells, and the mast cell in particular, in exercise-induced bronchoconstriction (EIB) has been inferred from studies showing a modification of the airway response by specific mediator antagonists 2 or the mast cell active drugs, such as cromoglycate and nedocromil 3.
Direct evidence for mediator release following exercise in asthmatics has been obtained by an increase in the urinary levels of the mast cell marker 9α,11β‐prostaglandin (PG)F2, a metabolite of PGD2, and leukotriene (LT)E4 in association with EIB 4–7.
A positive response to mannitol identifies asthmatics with EIB 8, 9 and there are no significant airway responses to mannitol in healthy nonasthmatics 10. Inhaled mannitol is thought to cause airways to narrow in asthmatics by a similar mechanism to exercise, that is by increasing the osmolarity of the airway surface liquid 8. It is known that mediator secretion from mast cells, basophils and human lung fragments is triggered by increased osmolarity 11, 12 and this release is inhibited with nedocromil sodium 13. As with EIB, the airway response to mannitol may be modified by specific mediator antagonists 14. However, there is no direct evidence for mediator release from mast cells or other cells occurring in response to inhaled mannitol.
The present study was undertaken to investigate if mannitol-induced bronchoconstriction in asthmatics was associated with mast cell activation and the release of mediators of bronchoconstriction, analogous to that observed in asthmatics with EIB. To this end the urinary levels of the PGD2 metabolite 9α,11β‐PGF2 were measured before and after a mannitol challenge in atopic asthmatics and in nonasthmatic nonatopic subjects who served as controls. The urinary excretion of Nτ‐methylhistamine was also measured, as well as LTE4, in view of pharmacological data supporting the role for cysteinyl-LTs and histamine in the bronchoconstriction following the inhalation of mannitol 14.
Methods
Subjects
Twelve atopic asthmatics (eight males, four females, aged 25–42 yrs) and nine nonatopic nonasthmatics (five males, four females, aged 22–33 yrs), all nonsmokers without any chest infection in the 4‐week period preceding the challenge, were included in the study (table 1⇓). Asthmatic subjects were required to have a baseline forced expiratory volume in one second (FEV1) >70% predicted while nonasthmatic subjects were required to have normal spirometry (all parameters) 15. To enter the study, it was a requirement for the asthmatic subjects that the reduction in FEV1 was sustained at ≥15% of baseline FEV1 for the 30 min following the challenge. Short-acting β2‐agonists were withheld for 6 h and nedocromil sodium or sodium cromoglycate was withheld for 48 h. Inhaled corticosteroids were not used on the day of the study. No subject was taking long-acting β2‐agonists or anti-histamines. All subjects were required to abstain from alcohol, caffeine and niacin-containing drinks and foods from 20:00 h the evening before the study and no vigorous exercise was permitted on the study day. The Central Sydney Area Health Service Ethics Committee approved the studies (Protocol No. X99-0089) and all subjects signed a consent form. The study was performed under the Clinical Trials Notification Scheme of the Therapeutic Goods Administration of Australia (CTN# 1999/150).
Anthropometric data, daily medication, dose of inhaled steroids, forced expiratory volume in one second (FEV1) % predicted, % reduction in FEV1, provoking dose of mannitol to cause a 15% fall in FEV1 (PD15) and total dose of mannitol for asthmatic and nonasthmatic subjects
Study design
At commencement, two baseline urine samples were collected 1 h apart, at 60 min before and immediately before the start of mannitol challenge, and spirometry measurements were performed. The FEV1 was used as the index of change in airway calibre (Microlab 3300 spirometer; Micro Medical, Kent, UK). A mannitol challenge was performed by progressively increasing the doses, as described previously 10. Briefly, the dose protocol consisted of 0 (empty capsule acting as a placebo), 5, 10, 20, 40, 80, 160, 160 and 160 mg mannitol. The 80- and 160‐mg doses were given in multiples of 40‐mg capsules. Three FEV1 manoeuvres wereperformed 60 s after each dose and the highest FEV1 measurement was recorded. The FEV1 value measured after the 0 mg capsule was used to calculate the per cent decrease in FEV1 in response to the mannitol challenge. If the subject had a decrease of ≥10% the dose producing this was repeated for safety reasons. The challenge was completed in asthmatic subjects when a ≥25% reduction in FEV1 was documented or, for the nonasthmatic subjects, when the maximum cumulative dose of mannitol was administered (635 mg). The provoking dose required to cause a 15% fall in FEV1 (PD15) for mannitol, an index of airway hyperresponsiveness, was calculated from the relationship between the per cent fall in FEV1 and the cumulative dose of mannitol required to provoke this fall. Airway recovery following mannitol was spontaneous and FEV1 was measured at 5 min and then at 10‐min intervals for up to 90 min after the start of the challenge.
Additional urine samples were collected at 30, 60 and 90 min after the challenge and all samples were stored without addition of preservatives at −80°C.
Mediator analyses
Enzyme immunoassay analyses of 9α,11β‐PGF2 and LTE4 were performed in unextracted urine samples with rabbit polyclonal antisera and acetylcholinesterase-linked tracers (Cayman Chemical Company, Ann Arbor, MI, USA) essentially as described previously 16. However, in the present study, a different antibody was used for analysis of LTE4, which cross-reacted with LTE4 to 67% and with LTC4 and LTD4 to 100%. Measurement of urinary Nτ‐methylhistamine was performed with a commercial double-antibody radioimmunoassay (Pharmacia AB, Uppsala, Sweden). Creatinine analyses were performed with a colorimetric assay (Sigma, StLouis, MO, USA).
Statistical analysis
All values are expressed as mean±sem unless otherwise stated. Mean values before and the peak value for concentration of urinary mediators (ng or µg·mmol creatinine−1) after challenge were compared by using the Student's t‐test and were considered significantly different if the p‐value was <0.05. Values were found to be normally distributed.
Results
Asthmatic subjects
All asthmatic subjects achieved the required sustained airway response following inhaled mannitol with a 34.5±2.1% reduction in FEV1 following a mean cumulative dose of mannitol of 272±56 mg (fig. 1⇓, table 1⇑). The airway response was associated with increases in the urinary excretion of 9α,11β‐PGF2 in all subjects (fig. 2a⇓), calculated as peak value versus mean of two baseline samples, with a mean significant increase (91.9±8.2 versus 66.9±6.6 ng·mmol creatinine−1; p<0.001). The levels of urinary 9α,11β‐PGF2 peaked at 30 min following the mannitol challenge, with levels back to baseline at 90 min (fig. 3a⇓).
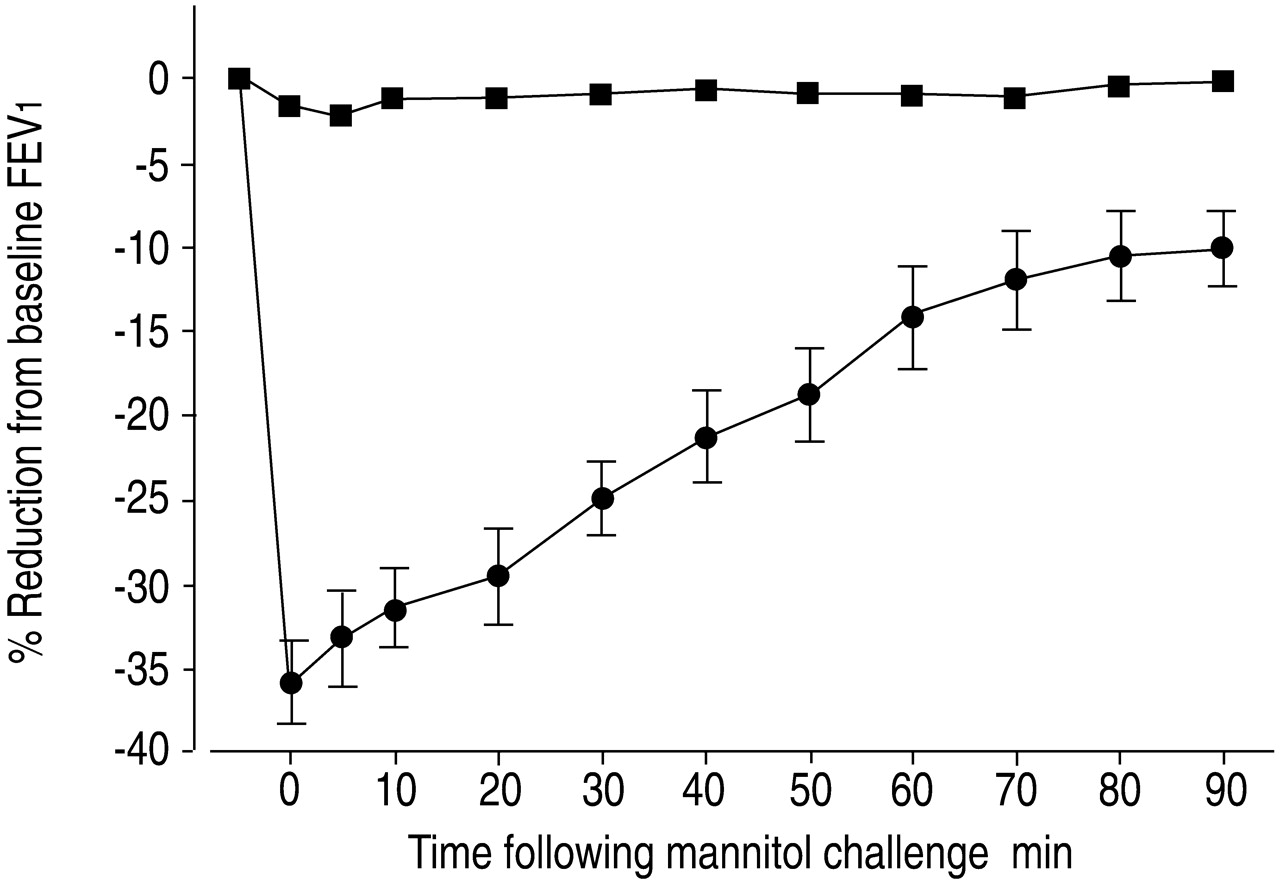
The percentage reduction in forced expiratory volume in onesecond (FEV1) from baseline following a mannitol challenge and spontaneous recovery in FEV1 in asthmatic (•: n=12) and nonasthmatic (▪: n=9) subjects. The mean±sem cumulative dose ofmannitol delivered to the asthmatic subjects was significantly lower (272±56 mg) compared with nonasthmatic subjects (635 mg) (p<0.001).

a) 9α,11β‐Prostaglandin (PG)F2, b) leukotriene (LT)E4 and c) Nτ‐methylhistamine in urine measured initially as the mean of the two baseline samples and then as the peak values measured over the 90 min collection period after the mannitol challenge in asthmatic (•) and nonasthmatic (○) subjects. Data are presented as mean±sem. ns: nonsignificant. *: p<0.05; **: p<0.01; ***: p<0.001.

{kind=link}
{kind=link}
{kind=link}
Mean±sem urinary excretion of 9α,11β‐prostaglandin F2 (•) and leukotriene E4 (○) in asthmatic subjects (a) and nonasthmatic subjects (b) over the 60 min collection period before and the 90 min period after the mannitol challenge.
There was also a significant increase in the urinary levels of LTE4 (51.3±7.5 versus 32.9±4.7 ng·mmol creatinine−1; p<0.05) (fig. 2b⇑). The post mannitol levels of LTE4 were sustained at 30–90 min, with no significant decline (fig. 3b⇑).
In contrast, there was no detectable increase in the urinary excretion of Nτ‐methylhistamine in response to the mannitol challenge (20.9±2.8 versus 18.2±2.1 µg·mmol creatinine−1; p=0.1) (fig. 2c⇑).
Nonasthmatic subjects
In nonasthmatic subjects there was no significant airway response to inhaled mannitol with a 1.0±0.5% reduction in FEV1 following challenge with the maximum cumulative doseof 635 mg (fig. 1⇑). This dose was significantly greater than the cumulative dose delivered to the asthmatic subjects (p<0.001). However, calculated as the peak versus mean of two baseline samples, there was a significant increase in theurinary excretion of 9α,11β‐PGF2 (68.4±6.9 versus 56.0±5.8 ng·mmol creatinine−1; p<0.05) (fig. 2a⇑). However, the magnitude of the increase in urinary excretion of 9α,11β‐PGF2 was less and the duration of the increased excretion was shorter than in the asthmatics, with values back to baseline at 60 min after challenge (fig. 3b⇑).
There was also a significant increase in urinary LTE4 (58.5±5.3 versus 43.0±3.3 ng·mmol creatinine−1; p<0.01) (fig. 2b⇑). The time-course for the urinary excretion of LTE4 was similar to the asthmatic subjects (fig. 3b⇑).
There was a small but significant increase in the urinary excretion of Nτ‐methylhistamine after challenge (21.6±2.8 versus 19.1±2.6 µg·mmol creatinine−1; p<0.05) (fig. 2c⇑).
Asthmatics versus nonasthmatics
There was no difference in the increase in LTE4 between nonasthmatic (15.4±3.3 ng·mmol creatinine−1) and asthmatic subjects (18.4±6.0 ng·mmol creatinine−1; p=0.68) (fig. 2b⇑). However, in the nonasthmatics the change in 9α,11β‐PGF2 (12.4±4.1 ng·mmol creatinine−1) was significantly smaller compared to asthmatic subjects (24.9±3.8 ng·mmol creatinine−1; p<0.05) (fig. 2a⇑). The changes in the levels of Nτ‐methylhistamine after mannitol were not different between asthmatics (2.6±1.5 µg·mmol creatinine−1) and nonasthmatics (2.5±0.8 µg·mmol creatinine−1; p=0.93) (fig. 2c⇑).
Baseline levels of urinary mediators did not differ between the asthmatic and nonasthmatic group of subjects; 9α,11β‐PGF2 (66.9±6.6 versus 56.0±5.2 ng·mmol creatinine−1; p=0.25), LTE4 (32.9±4.7 versus 43.0±3.3 ng·mmol creatinine−1; p=0.12) and Nτ‐methylhistamine (18.2±2.2 versus 19.1±2.6 µg·mmol creatinine−1; p=0.80) (fig. 2a–c⇑).
Discussion
The results of this study demonstrate that the airway narrowing provoked by challenge with inhaled mannitol in asthmatic subjects is associated with increased urinary excretion of 9α,11β‐PGF2. The parent compound PGD2 is almost exclusively released by mast cells upon activation and is subsequently enzymatically converted to 9α,11β‐PGF2 in sites such as the lungs 7. This compound is, in turn, excreted into the urine as a stable and dominating metabolite, and increased levels may be regarded as an index of mast cell activation 7. Therefore, it is concluded that mannitol caused activation of mast cells in the airways of the asthmatics and this is probably a consequence of mannitol causing an increase in osmolarity of the airway surface liquid. Moreover, the increased excretion of the PGD2 metabolite may indicate an important role for PGD2 in the bronchoconstrictive response to mannitol. PGD2, as well as 9α,11β-PGF2, are potent bronchoconstrictors in man 17, mainly acting on the TP‐receptor 18, 19. Pretreatment with selective TP‐receptor antagonists attenuated both PGD2- and allergen-induced, but not histamine-induced, bronchoconstriction 20–22. Release of PGD2 into the airways has been further documented after bronchial provocation with allergen 23. Interaction between cys-LTs and PGD2 in provoking bronchoconstriction has also been indicated in asthmatic subjects 24.
The finding that the mannitol challenge only caused a minor change in urinary excretion of Nτ-methylhistamine may seem to contradict the conclusion about mast cell activation. However, the current authors have previously observed that urinary methylhistamine is a much less sensitive marker of mast cell activation in vivo. For example, following allergen challenge, the increase in 9α,11β‐PGF2 was much greater than that of methylhistamine 16. Of particular interest to this study, is the previous finding that EIB was also associated with significantly increased urinary 9α,11β‐PGF2 6, 7 at a time when only a nonsignificant trend for increased urinary Nτ‐methylhistamine was found 6. Mannitol is known to induce the release of histamine from lung mast cells in vitro 12. Furthermore, the histamine antagonist, fexofenadine, is effective in reducing airway sensitivity to mannitol 14. This suggests that preformed histamine is involved in the response to mannitol and its source is likely to be mast cells. Whilst basophils are also a source of histamine they do not synthesise PGD2 25. Nedocromil sodium inhibits the airway response to mannitol 26 and this effect is thought to occur by inhibiting or preventing the release of avariety of mast cell mediators 13. The finding that there was no significant increase in urinary excretion of Nτ‐methylhistamine in the asthmatic subjects suggest that the provoking stimulus in this group was insufficient to cause an increase in systemic histamine large enough to be reflected in urine.
It was also found that the mannitol-induced bronchoconstriction was associated with increased urinary LTE4 excretion. The finding is consistent with the previous demonstration that the LT antagonist montelukast inhibited the airway response to mannitol, as demonstrated by a faster recovery of the lung function to baseline after challenge 14. Moreover, the observations add to the similarities with EIB, as it is well established that recovery of lung function to baseline after EIB is also much faster with antagonists of the cysteinyl‐LT 2, 5, 27, 28. Measurements of urinary LTE4 have also demonstrated increased levels after EIB 4, 5, although the magnitude is smaller and less consistent than after allergen-induced bronchoconstriction. As a technical note, it should be recognised that the time course of the bronchoconstriction after exercise and mannitol is short and this makes it very different to the prolonged airway responses that occur after allergen challenge. This may explain why it has been difficult to find significantly increased levels of urinary LTE4 after exercise in some studies 6. It has been documented that the duration of exposure to a stimulus that causes bronchoconstriction determines the magnitude of overflow of LTE4 into the urine 29. The similar profiles of mediator excretion into the urine after mannitol and exercise in asthmatics provide evidence for mast cell activation as a common mechanism for the airway narrowing.
An unexpected finding was that the inhalation of 635 mg mannitol by the nonasthmatic subjects was also associated with a significant increase in levels of LTE4 and 9α,11β‐PGF2 and small changes in Nτ‐methylhistamine. However, the cumulative dose of 635 mg mannitol inhaled by the nonasthmatics was between one to 18 (median three)-times the dose inhaled by the asthmatics. Furthermore, with this stronger stimulus for mast cell activation, the increased urinary excretion of 9α,11β‐PGF2 in the nonasthmatic subjects was only about half that compared with the asthmatics and of shorter duration with levels back to baseline at 60 min after challenge. These findings suggest that the mast cells of the asthmatics were easier to activate or more numerous, or both. In keeping with this, a recent study reports that asthmatic subjects have significantly higher numbers of mast cells in the airways and a particularly higher proportion localised in the smooth muscle compared to nonasthmatic subjects 30. In addition, eosinophils are more abundant in the airways of asthmatics with ongoing airway inflammation compared to nonasthmatics 30. The cellular source of the LTs cannot be established in this study and although the mast cell is the most likely cell source, other cells, such as eosinophils, could have been involved.
As expected the nonasthmatic subjects did not have a reduction in FEV1 despite having significantly increased urinary levels of LTE4 and 9α,11β‐PGF2, which, as well as its parent compound PGD2, is a potent bronchoconstrictor 17. The major difference between the response to inhaled mannitol in the two groups studied is most probably related to differences in airway responsiveness to the released mediators. It has been extensively documented that asthmatics have more sensitive airways than nonasthmatics and that airway narrowing would be expected to occur at low concentrations of these mediators 31. A measurement of airway responsiveness to inhaled PGs or LTs was not part of this protocol, but hyperresponsiveness to PGD2, as well as LTD4, in asthmatics has been reported previously 32, 33. Interestingly, in line with the present authors' hypothesis, it was documented that subjects presenting with EIB were hyperreactive to PGD2 34.
From the findings presented here with mannitol challenge, it is suggested that the difference between asthmatic and nonasthmatic subjects is in the airway responsiveness to mediators of bronchoconstriction. This factor distinguishes the two groups more than differences in the degree of release of inflammatory mediators. This theory does not preclude the fact that there may also be differences with respect to the ease by which mediators can be released, but such discrepancies may be of comparatively less importance.
In conclusion, this study has documented that mannitol challenge is associated with mast cell activation and release of mediators of bronchoconstriction in asthmatic as well as in nonasthmatic subjects. This supports the proposal that increased osmolarity of the airway fluid lining is a general stimulus for activation of mast cells. Similar time courses of the urinary excretion of 9α,11β‐prostagladin F2 were seen after exercise 6 and mannitol (present study). These findings lend further support to the concept that the stimulus and mechanism of airway narrowing to mannitol and exercise are similar 8, 9. The authors also conclude that mannitol causes bronchoconstriction only in the asthmatics, because only the asthmatic subjects display increased bronchial responsiveness to the liberated mediators. Future studies with combinations of different selective pharmacological interventions may further delineate the relative importance of the different mediators in causing the bronchoconstriction elicited by osmotic challenge. In particular, studies with agents blocking the biosynthesis or receptor-mediated action of prostglandin D2 are implicated based on the findings in the present study.
Acknowledgments
The authors would like to thank B. Watson of the Dept of Molecular Genetics, RPA Hospital, Camperdown, Australia, for his help in storing the samples and C. Perry for her technical assistance in preparing the manuscript. They would also like to thank S‐E. Dahlén for his helpful discussions in the design and analysis of the results. The use application for mannitol described in this study is covered by United States of America Patent No. 5,817,028 and internationally by PCT/AU95000086. The patent is owned by the Central Sydney Area Health Service and is licensed to Pharmaxis Pty Ltd NSW Australia.
- Received January 30, 2003.
- Accepted April 9, 2003.
- © ERS Journals Ltd
References









