





Abstract
Primary pulmonary hypertension (PPH) and Castleman's disease (CD) are rare conditions infrequently encountered in clinical practice.
In this paper, two patients diagnosed with both of these diseases are reported. The authors speculate that rather than being a chance occurrence, these conditions are linked by a common angio-proliferative mechanism. Therefore, an association between infection with the human herpesvirus‐8 and the diseases of PPH and CD was sought.
Evidence of human herpesvirus‐8 infection was found in the lung tissue and, specifically, in the plexiform lesions from one of the patients.
Primary (P) or unexplained pulmonary hypertension (PH) is a poorly understood disease with devastating clinical consequences. Patients with PPH develop a marked and unexplained elevation of pulmonary artery pressure that often results in right heart failure and death 1.
The plexiform lesion is one of the characteristic histopathological lesions of PPH. Recent investigation has emphasised the important role of endothelial cells (ECs) in the disease process of severe PH 2. The plexiform lesion itself is composed of a disorganised “tumour like” proliferation of endothelial cells. In patients with PPH the ECs that compose the plexiform lesion are monoclonal in origin while in patients with secondary PH they are polyclonal 3, 4. The ECs of the plexiform lesions in PPH patients possess microsatellite instability and abnormal expression of growth and apoptosis control genes similar to some forms of neoplasia 5.
Castleman's disease (CD) is a highly heterogeneousclinical-pathological entity belonging to the lymphoproliferative disorders 6. Histologically CD is classified into two major types according to pathological findings: 1) the hyaline-vascular type with small hyaline-vascular follicles and interfollicular capillary proliferation; and 2) the plasma-cell type characterised by a massive accumulation of polyclonal plasma-cells in the interfollicular region. There is also a third, mixed variant, with features of both hyaline vascular and plasma-cell types 6, 7. Clinically, CD can be classified into either localised or systemic forms. The usual presentation for localised disease is a single enlarged lymph node or widened mediastinum. The systemic form of the disease, also referred to as multicentric CD (MCD), is characterised by diffuse lymphadenopathy, hepatosplenomegaly and constitutional symptoms. Localised CD of the hyaline vascular type is usually asymptomatic, whereas patients with the plasma cell variant often have systemic manifestations. Patients with MCD have significant lymphadenopathy and an aggressive, sometimes fatal, clinical course 6. Although the aetiology ofCD is unknown, levels of cytokines such as interleukin (IL)‐1, ‐6 and tumour necrosis factor‐β are increased in the serum of patients with this disease 8. Others 7 have reported an increased expression of vascular endothelial growth factor (VEGF) in the serum and lymph nodes of patients with CD. VEGF is a specific mitogen for vascular endothelial cells and plays a central role in angiogenesis. Nishi and Maruyama 7 hypothesised that the marked vascular proliferation in the interfollicular space seen in this disease process was related to the elevated VEGF levels. Interestingly, it has been documented that patients with PPH have increased levels of VEGF in their serum and the plexiform lesions of PPH express high levels of this growth factor and its receptor 9, 10. It has been hypothesised that the increased serum VEGF levels found in patients with PPH serve as a stimulus for endothelial cell proliferation, perhaps contributing to the pathobiology of the disease 3.
Recently, human herpesvirus (HHV)‐8 has been associated with MCD and a viral homologue of IL‐6 (vIL‐6) encoded by HHV‐8 has been demonstrated to induce VEGF expression 7, 11. Viral infection with agents such as human immunodeficiency virus type‐1 (HIV‐1) is associated with the development of severe PH, indistinguishable clinically or pathologically from PPH. Although a direct causal relationship between PH and a viral infection has not been established, such a relationship is strongly suspected 12, 13.
POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes) syndrome is a rare variant of plasma cell dyscrasia with multiple systemic manifestations 14. POEMS syndrome has been associated with both MCD and with HHV‐8 infection 11, 15. Intriguingly, severe PH, with histology similar to that found in PPH (plexiform lesions and in situ micro thrombosis) has been reported in patients with POEMS syndrome 16. It has also been noted that patients with POEMS syndrome and PH have elevated serum levels of VEGF 16.
In this paper, two patients diagnosed with both PPH and MCD are reported. An evaluation for an association between PPH, CD and HHV-8 was undertaken in these two patients.
Case 1
A 34‐yr-old Hispanic female (patient 1) presented with the chief complaint of a nonproductive cough, increasing dyspnoea and new-onset lower extremity oedema. The patient denied a previous history of deep venous thrombosis, pulmonary embolism, exposure to anorexic agents or intravenous drug use. An HIV test was negative. There was no evidence of a connective tissue disease on physical exam. An accentuated pulmonic component of the second heart sound was audible but lung sounds were normal. A chest radiograph demonstrated prominent pulmonary arteries and right ventricular hypertrophy. The patient's electrocardiogram was consistent with right ventricular hypertrophy. An echocardiogram demonstrated normal left ventricular systolic function. The right atrium and right ventricle were dilated and severe tricuspid regurgitation was noted. The pulmonary artery systolic pressure was estimated to be 60–65 mmHg plusright atrial pressure. A ventilation perfusion scan was interpreted as low probability for pulmonary embolism. A high resolution computed tomography (CT) scan of the patients chest demonstrated normal lung parenchyma but enlargement of the pulmonary arteries. Pulmonary function tests demonstrated mild airflow limitation and a decreased carbon dioxide diffusing capacity of the lung (DL,CO, 58%) (DL,CO/alveolar volume (VA) 72%). A sleep study demonstrated no evidence of obstructive sleep apnoea. Blood serology for connective tissue disease demonstrated a positive antinuclear antibody of 1:256 with a speckled pattern. There was no other evidence of collagen vascular disease. Other markers of connective tissue disease were normal. Right heart catheterisation with a vasodilator trial (table 1⇓) did not demonstrate a “significant” acute response to the vasodilator prostacyclin. The patient was diagnosed with severe, unexplained (P) PH and continuous i.v. prostacyclin therapy was initiated. The patient also began evaluation for lung transplantation, during which a pelvic and abdominal CT scan revealed a 4.0×6.0×3.5 cm abdominal mass (fig. 1⇓). The patient underwent laparotomy with removal of the mass, seemingly in its entirety. Histological evaluation demonstrated the plasma-cell variant of CD (fig. 2⇓). The patient was initially followed conservatively, but in the ensuing months was noted to have increased lymphadenopathy and the development of weight loss and night sweats. The patient was given a trial of cytoxan and decadron without noticeable improvement. The patient was then treated with four weekly doses of rituxan. This resulted in a complete resolution of all lymphandenopathy and “B” type symptoms. The patient was judged to be in complete remission from CD although severe PH persisted.

Computed tomographic scan of the abdomen of patient 1, demonstrating mass lesion (arrow).

Lymph node with evidence of Castleman's disease from patient 1. Scale bar=100 µm.
Cardiac catheterisation results from patients 1 and 2 at baseline and at maximum infused dose of prostacyclin (max. PGI2)
Three years after the diagnosis of PPH and the initiation of continuously infused prostacyclin, the patient was admitted for increasing shortness of breath, hypoxia and rapidly increasing lower extremity oedema. Careful diuresis was attempted but the patient suffered a bradycardic arrest during the hospitalisation. Aggressive attempts at resuscitation were unsuccessful and the patient expired 5 days after admission. The cause of death was determined to be severe PH and right heart failure. At autopsy no residual evidence of CD was uncovered but examination of the lung tissue confirmed the presence of plexogenic pulmonary arteriopathy (fig. 3⇓).

Plexiform lesion from patient 1 (arrow). Scale bar=100 µm.
Case 2
A 44‐yr-old Caucasian male (patient 2) was referred for evaluation of progressively worsening lower extremity oedema and increasing dyspnoea. The patient denied a history of appetite suppressant use or connective tissue disease. Blood serology for HIV was negative. The patient had a history of obstructive sleep apnoea, which was being treated successfully with continuous positive airway pressure. The patient denied any previous history of deep venous thrombosis or pulmonary embolism. Physical exam demonstrated a prominent second heart sound and an III/VI systolic murmur loudest at the left upper sternal border. The patient was also noted to have 2+ bilateral lower extremity oedema and both anterior-cervical and inguinal lymphandenopathy. A chest radiograph demonstrated enlargement of the cardiac silhouette. An echocardiogram demonstrated normal left ventricular wall thickness and function. The right atrium was dilated and there was moderate tricuspid regurgitation. The right ventricular systolic pressure was estimated to be 45 mmHg. A ventilation-perfusion scan was interpreted as low probability for pulmonary embolus. Bilateral lower extremity ultrasound demonstrated no evidence of deep vein thrombosis. Pulmonary function tests demonstrated normal spirometry, mildly decreased lung volumes thought to be related to the patient's body habitus and a decreased DL,CO (47%) (DL,CO/VA 72%). Physical exam and serology did not reveal evidence of a connective tissue disease.
The patient underwent right heart catheterisation with a trial of the vasodilator prostacyclin (table 1⇑). The diagnosis of unexplained (P) PH was made. The patient demonstrated a significant response to prostacyclin with a 15% decrease in mean pulmonary artery pressure (Ppa) and >25% decrease in pulmonary vascular resistance. During the same hospitalisation the patient was noted to have nocturnal fevers and night sweats. Physical exam demonstrated anterior cervical and supraclavicular adenopathy. A CT scan of the neck, chest and abdomen confirmed significant supraclavicular, pre-tracheal and bilateral inguinal adenopathy. The patient underwent excision of a right supraclavicular lymph node. The biopsy demonstrated a mixed variant of CD (fig. 4⇓). The patient was initially started on a calcium channel blocker for treatment of PH, however, the patient was intolerant of this medication and was changed to the endothelin antagonist bosentan. Other treatment consisted of four weekly doses of rituxan for CD. Eight months after starting these medications the patient's lymphandenopathy resolved and Ppa was significantly decreased.

Lymph node from patient 2 demonstrating a regressively transformed germinal center. Hyalinised capillaries are evident (arrow). Scale bar=100 µm.
Evaluation for an association between PPH, CD and HHV‐8 was undertaken in these two patients.
Materials and methods
Specimens
The specimens were obtained from lymph node and lung autopsy samples from patient 1 and from a lymph node biopsy of patient 2. Positive controls for polymerase chain reaction (PCR) and immunohistochemistry were selected from other patient specimens previously diagnosed with Kaposi's sarcoma. Negative controls were from lung and lymph node sections of patients with unrelated diagnoses. Allsamples were prepared from formalin-fixed, paraffin-embedded tissue sections.
Immunohistochemistry
The primary antibody used was a rat immunoglobulin G1 to open reading frame (ORF) 73 (Advanced Biotechnologies Inc, Columbia, MD, USA). Biotinylated secondary antibodies were from the Vectastain Elite Rat ABC Kit (Vectastain, Burlingame, CA, USA) as per the manufacturer's recommendations.
Polymerase chain reaction
PCR was performed on lung and lymph node tissue using the primers for ORF 72 (vCyclin) and the protocol as described by Pan et al. 17. The primers used were: forward (5′‐CGCCTGTAGAACGGAAACAT‐3′); and reverse (5′‐TTGCCCGCCTCTATTATCAG‐3′) with a 138 base pair product. The PCR reaction mixture consisted of 25 mM MgCl2, 10 mM dNTPs, 1 µM of each primer, 5 U·mL−1 Taq polymerase (Boehringer Mannheim, Indianapolis, IN, USA), in a total volume of 25 µL. Amplification was performed in an oil-free thermocycler (Model GeneAmp 2400; Perkin Elmer, Foster City, CA, USA). The thermocycler conditions were 94°C for 2 min, 94°C for 1 min, 58°C for 1 min and 72°C for 1 min for 35 cycles, then 72°C for 5 min.
PCR was performed on serum samples of patient 2, evaluating the HHV‐8 minor capsid (ORF 26), from nucleotides 47311 to 47384 of the HHV‐8 genome, as described previously by White and Campbell 18. Deoxyribonucleic acid (DNA) sequencing was performed in the University of Colorado School of Medicine DNA sequencing and analysis core using ABI Prism kits from Applied Biosystems (Foster City, CA, USA).
Enzyme-linked immunosorbent assay
The VEGF level present in the serum of patient 2 was quantified by enzyme-linked immunosorbent assay (ELISA) (R&D Systems Inc., Minneapolis, MN, USA).
Indirect immunofluorescense assay
The presence of antibodies to Kaposi's sarcoma-associated herpesvirus latent nuclear antigens (LANA) was determined by immunofluorescent assay (IFA) with latency infected BCP‐1 cells as previously described 19.
Results
The immunohistochemistry of the lung sections from patient 1 was positive for HHV‐8 (fig. 5⇓). The lymph node sections from patients 1 and 2 were negative for HHV‐8 by staining (data not shown).
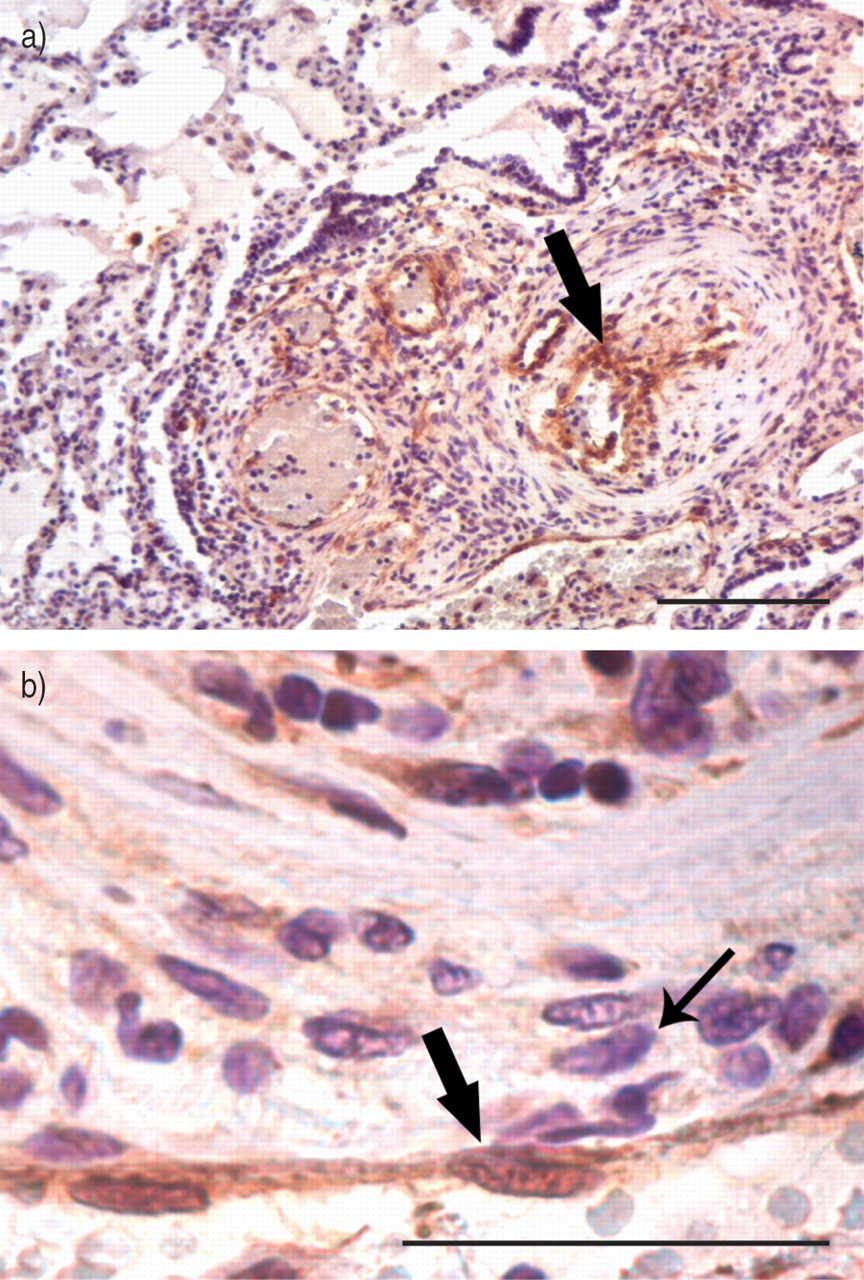
Tissue section from patient 1 (lung) demonstrating positive staining for human herpesvirus (HHV)‐8. a) The cells that stained positive for HHV‐8 were predominantly endothelial cells and lymphocytes located within and around the plexiform lesion (large arrow). Scale bar=100 µm. b) Positive staining of endothelial cells (large arrow). Note the negative staining of the smooth muscle cells (small arrow). Lymph nodes from patients 1 and 2 were negative for HHV‐8 by immunohistochemistry (data not shown). Kaposi's Sarcomalesions served as positive controls (data not shown). Scale bar=50 µm.
The PCR of lung tissue from patient 1 was positive for HHV‐8 using the primers for ORF 72. The PCR product was sequenced and was shown to be 97% homologous to the expected product for ORF 72 (data not shown). The lymph nodes from patients 1 and 2 were negative by PCR for HHV‐8 (fig. 6⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Polymerase chain reaction for human herpesvirus (HHV)‐8 (ORF72). Lane 1 is a positive control using a Kaposi's sarcoma (KS) lesion. Lane 2 is from lung tissue of patient 1 (Pt 1 lung) (positive). Lane 3 is lymph node tissue from patient 1 (Pt 2 node) (negative). Lane 4 is lymph node tissue from patient 2 (Pt 2 node) (negative).
A serum sample from patient 2 was negative for HHV‐8 by PCR for the minor capsid (ORF 26). The amount of VEGF present in his peripheral blood as measured by ELISA was within normal limits (118 pg·mL−1). Serology from patient 2 was negative for HHV‐8 by IFA.
Discussion
PPH and CD are both rare pathological conditions. The occurrence of both diseases in two different patients is remarkable and raises interesting questions regarding pathogenesis. Both CD and PPH have been associated with high serum VEGF levels and both diseases have been described as angioproliferative and vasculoproliferative processes 3, 7. The current authors hypothesised that HHV‐8 might be a common inciting agent for both diseases in the two patients presented here. There was immunohistochemical evidence of HHV‐8 infection in the lung tissue of patient 1, although there was no evidence in the lymph nodes of either patient. Importantly, the HHV‐8 staining was noted in the endothelial cells in and around the plexiform lesion while other cells, such as those of smooth muscle, were negative. HHV‐8 commonly infects endothelial cells 20. There was also PCR evidence of HHV‐8 in the lung tissue of patient 1, but again not in the lymph nodes of either patient. In the serum of patient 2, there was no PCR evidence of active HHV‐8 infection, the VEGF levels were not elevated and there was no positive serology for HHV‐8 (these tests could not be performed on patient 1 as the patient had expired by this time). However, it should be noted that the serum HHV‐8 PCR and VEGF levels were drawn late in the patient's course and after the initiation of treatment with resolution of the patient's lymphandenopathy and significant improvement of PH. It should also be noted that not all patients with HHV‐8 infection have positive serology. The reported sensitivity for HHV‐8 LANA seropositivity in patients with Kaposi's sarcoma is only 70–80% 21, 22.
The potential role of HHV‐8 in the development of PH is intriguing. The authors speculate that HHV‐8 might induce the production of VEGF in the infected host via a viral homologue of IL‐6 and this might result in an angiogenic cascade, as proposed initially by Nishi and Maruyama 7. If the HHV‐8 infection was local, for example in the lungs or the lymph nodes, this could explain the organ-specific features ofboth PPH and CD. The current authors were unable to demonstrate an elevated VEGF level in patient 2, but elevated VEGF levels in PPH and CD have been reported previously 7, 9.
It has been speculated that primary pulmonary hypertension may occur in individuals with a genetic predisposition for the disease who are then exposed to a “second hit” resulting in changes in the proliferation of pulmonary vascular endothelial cells and the development of severe pulmonary hypertension. Here, the authors suggest that infection with human herpesvirus‐8 may serve as a second hit in some cases. This hypothesis now needs to be tested in a larger cohort of patients.
- Received January 19, 2003.
- Accepted April 17, 2003.
- © ERS Journals Ltd
References









