





Abstract
The efficacy and safety of budesonide/formoterol in a single inhaler compared with placebo, budesonide and formoterol were evaluated in patients with moderate-to-severe chronic obstructive pulmonary disease (COPD).
In a 12-month, randomised, double-blind, placebo-controlled, parallel-group study in 812 adults (mean age 64 yrs, mean forced expiratory volume in one second (FEV1) 36% predicted normal), patients received two inhalations twice daily of either budesonide/formoterol (Symbicort®) 160/4.5 µg (delivered dose), budesonide 200 µg (metered dose), formoterol 4.5 µg or placebo. Severe exacerbations and FEV1 (primary variables), peak expiratory flow (PEF), COPD symptoms, health-related quality of life (HRQL), mild exacerbations, use of reliever β2‐agonist and safety variables were recorded.
Budesonide/formoterol reduced the mean number of severe exacerbations per patient per year by 24% versus placebo and 23% versus formoterol. FEV1 increased by 15% versus placebo and 9% versus budesonide. Morning PEF improved significantly on day 1 versus placebo and budesonide; after 1 week, morning PEF was improved versus placebo, budesonide and formoterol. Improvements in morning and evening PEF versus comparators were maintained over 12 months. Budesonide/formoterol decreased all symptom scores and use of reliever β2‐agonists significantly versus placebo and budesonide, and improved HRQL versus placebo. All treatments were well tolerated.
These results suggest a role for budesonide/formoterol in the long-term management of moderate-to-severe chronic obstructive pulmonary disease.
This study was supported by AstraZeneca.
Chronic obstructive pulmonary disease (COPD) is currently the fourth leading cause of death in the world 1, with increasing prevalence and mortality predicted in the coming decades 2. COPD is a serious and disabling disease, which imposes a large burden on patients, healthcare systems and society.
In patients with COPD, lung function deteriorates progressively over several years with increasing symptoms (e.g. dyspnoea, chest tightness, cough and sputum production); acute exacerbations are common, particularly in later stages, and these have considerable impact on patients' daily activities and well-being 3. Cigarette smoking is the major aetiological factor in COPD and smoking cessation is the only factor which has been shown to influence the decline in forced expiratory volume in one second (FEV1) 4, 5. However, the COPD-related inflammatory process in the airways initiated by smoking persists after cessation of smoking 6, and effective treatment is needed in past smokers with COPD 7.
The pharmacotherapy of COPD largely consists of mucolytics, bronchodilators, such as β2‐agonists, anticholinergics, theophylline and anti-inflammatory drugs i.e. inhaled corticosteroids, often taken in combination 2. Consequently, there is a need for better treatment options to relieve symptoms, reduce exacerbations and to provide better health-related quality of life (HRQL) for individual patients.
The long-acting β2‐agonists formoterol and salmeterol have been found to improve lung function and reduce both symptoms and the need for rescue medication in COPD compared with other treatments 8. Studies have also shown that formoterol and salmeterol can improve exercise tolerance 9, 10 and HRQL 11, 12 in patients with COPD compared with other treatments. The unique rapid- and long-acting properties of formoterol, however, give this treatment a faster onset of effect 13, similar to salbutamol 14.
Treatment with inhaled corticosteroids has shown clinical benefits in COPD by improving exacerbations, symptoms, lung function and HRQL 15–18. Sin and Tu 19 observed that elderly patients recently hospitalised for COPD who had received inhaled corticosteroid therapy following discharge from hospital had 24% fewer repeat hospitalisations for COPD, while 29% of these patients were less likely to die during 1 yr of follow-up.
In view of these effects, and since complementary actions of β2‐agonists and corticosteroids have been shown both in vitro 20, 21 and in patients with asthma 22, 23, this study was designed to evaluate the efficacy and safety of budesonide/formoterol in a single inhaler (Symbicort®; AstraZeneca, Lund, Sweden) against placebo and both monocomponents in patients with moderate-to-severe COPD.
The primary efficacy variables were number of severe exacerbations and FEV1. Secondary efficacy variables comprised vital capacity (VC), HRQL and diary card data: COPD symptoms, peak expiratory flow (PEF), use of reliever medication and mild exacerbations. The secondary objective was to evaluate safety, through adverse event (AE) reporting, electrocardiography, haematology and clinical chemistry.
Methods
Study patients
Adults with moderate-to-severe COPD (in line with Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines for the diagnosis, management and prevention of COPD) 2 were included and selected according to the following criteria: out-patients aged ≥40 yrs; COPD symptoms for ≥2 yrs; ≥10 pack-yrs smoking history; FEV1/VC ≤70%; FEV1 ≤50% predicted normal (stages IIB and III according to the GOLD classification) 2; total symptom score ≥2 per day during at least 7 days of the run-in period; documented use of short-acting inhaled bronchodilators for reliever medication; ≥1 severe COPD exacerbation within 2–12 months before the first clinic visit.
Patients were excluded if they had a history of asthma and/or seasonal allergic rhinitis before the age of 40, had any relevant cardiovascular disorders as judged by the investigator, were using β‐blocking agents, had current respiratory tract disorders other than COPD or any other significant diseases or disorders which may have put them at risk or which may have influenced the results of the study, had a requirement for regular use of oxygen therapy or had an exacerbation during run-in. Patients for whom it would have been considered unethical to withdraw inhaled steroids were also excluded. All patients gave written, informed consent and the study was approved by Ethics Committees for each centre.
Demographic and baseline characteristics were similar across all treatment groups; mean age was 64 yrs, mean baseline FEV1 was 0.99 L (36% pred) and mean smoking history was 44 pack-yrs (table 1⇓).
Patient demographic and baseline characteristics
Study design
This was a randomised, double-blind, placebo-controlled, parallel-group, multicentre study involving 89 centres from 11 countries (Argentina, Brazil, Denmark, Finland, UK, Italy, Mexico, Poland, Portugal, South Africa and Spain).
Preventative medication was withdrawn from all potential subjects prior to recruitment (visit 1): parenteral steroids, oral steroids, antibiotics and nebulised treatment from 4 weeks before; inhaled steroids from 2 weeks before; inhaled long-acting β2‐agonists from 48 h before; inhaled short-acting β2‐agonists from 6 h before; other bronchodilators from 6–48 h before.
Visit 1 was followed by a 2‐week run-in period; patients were issued with terbutaline 0.5 mg for relief but no other therapy was allowed during run-in. At visit 2, patients were randomised into groups for 12 months treatment. Further clinic visits took place at 1, 2, 3, 6, 9 and 12 months (visits 3–8, respectively). Only study medication was allowed during the treatment period, plus terbutaline 0.5 mg when needed as reliever medication.
Treatments
Treatment consisted of two inhalations of either budesonide/formoterol (Symbicort®) 160/4.5 µg (total delivered dose 320/9 µg), budesonide (Pulmicort®) 200 µg metered dose, formoterol (Oxis®) 4.5 µg delivered dose or placebo, all given twice daily. Terbutaline 0.5 mg per inhalation (metered dose) (Bricanyl® Turbuhaler®) was allowed throughout the study, when needed, as reliever medication for all patients, including the placebo arm.
Assessments
Severe exacerbations (use of oral steroids and/or antibiotics and/or hospitalisation due to respiratory symptoms) were recorded at visits 3–8 in each patient's case report form. Patients used daily diary cards to record their morning and evening COPD symptoms (shortness of breath, cough, chest tightness and night-time awakenings; each symptom scored 0–4, none to severe/almost constant), their short-acting β2‐agonist use (reliever medication), use of other COPD medication and PEF. PEF measurements were taken before the morning and evening doses of study medication. Mild exacerbations (a day with ≥4 inhalations of reliever medication above the mean run-in use) were calculated from daily diary card data. Spirometry was performed at each clinic visit, after study medication and at least 6 h after the last use of reliever medication.
A health status questionnaire, the St. George's Respiratory Questionnaire (SGRQ) 24 was completed at visits 1 (for training purposes), 2, 6 and 8. SGRQ total scores and scores from the three domains (activity, symptoms, impact) were calculated.
Patients were asked a standard question to detect AEs at visits 2–8; haematology, clinical chemistry and electrocardiography (ECG) were carried out at visits 1, 6 and 8.
Analysis
Assuming a mean of one exacerbation per patient per year, 150 patients per group would give 80% power to detect a reduction of 25% in the rate of severe exacerbations between two groups. For FEV1, 150 patients per group would give 80% chance of detecting a difference of 5% between two treatments (assuming a residual sd of 0.15 units on the logarithmic scale).
An intention-to-treat analysis was used and all hypothesis testing was with two-sided alternative hypotheses; p<0.05 was considered statistically significant. Severe (and mild) exacerbations and oral steroid courses, expressed as mean rate (mean number of exacerbations per patient per year), were analysed using a Poisson regression model; treatment and country were used as factors, time in study as an offset variable and confidence intervals were adjusted for overdispersion. Rate ratios from this model were presented as per cent reductions. The FEV1 end-point was the mean of all available measurements during the treatment period, analysed in a multiplicative analysis of variance (ANOVA with logarithm of values) with factors, treatment, country and visit 2 value as covariates. The mean ratios were presented as per cent increases. It was required that both primary variables should give statistical significance at the 5% level in order to keep the overall significance level to 5% in the final conclusion 25. Diary card variables were analysed in a similar manner to FEV1 but with an additive model. SGRQ was also similarly analysed but based on the last available measurement on treatment.
Results
Patient flow
Nine hundred and eighty patients were enrolled, of whom 168 were withdrawn prior to randomisation, the largest number (45% of the total) due to deterioration of COPD symptoms. A total of 812 patients were randomised, of whom 537 completed the study. There were 275 withdrawals following randomisation, with 173 due to AEs; 115 of these were due to COPD deterioration, the highest number occurring in the placebo group (table 2⇓). There were fewer withdrawals in total and due to COPD deterioration in the active treatment groups compared with the placebo group (for both comparisons budesonide/formoterol versus placebo p<0.001, budesonide and formoterol versus placebo p<0.05). A Kaplan-Meier plot of discontinuations shows that the probability of still remaining in the study on a particular day was higher for all active treatment groups compared with placebo (fig. 1⇓).
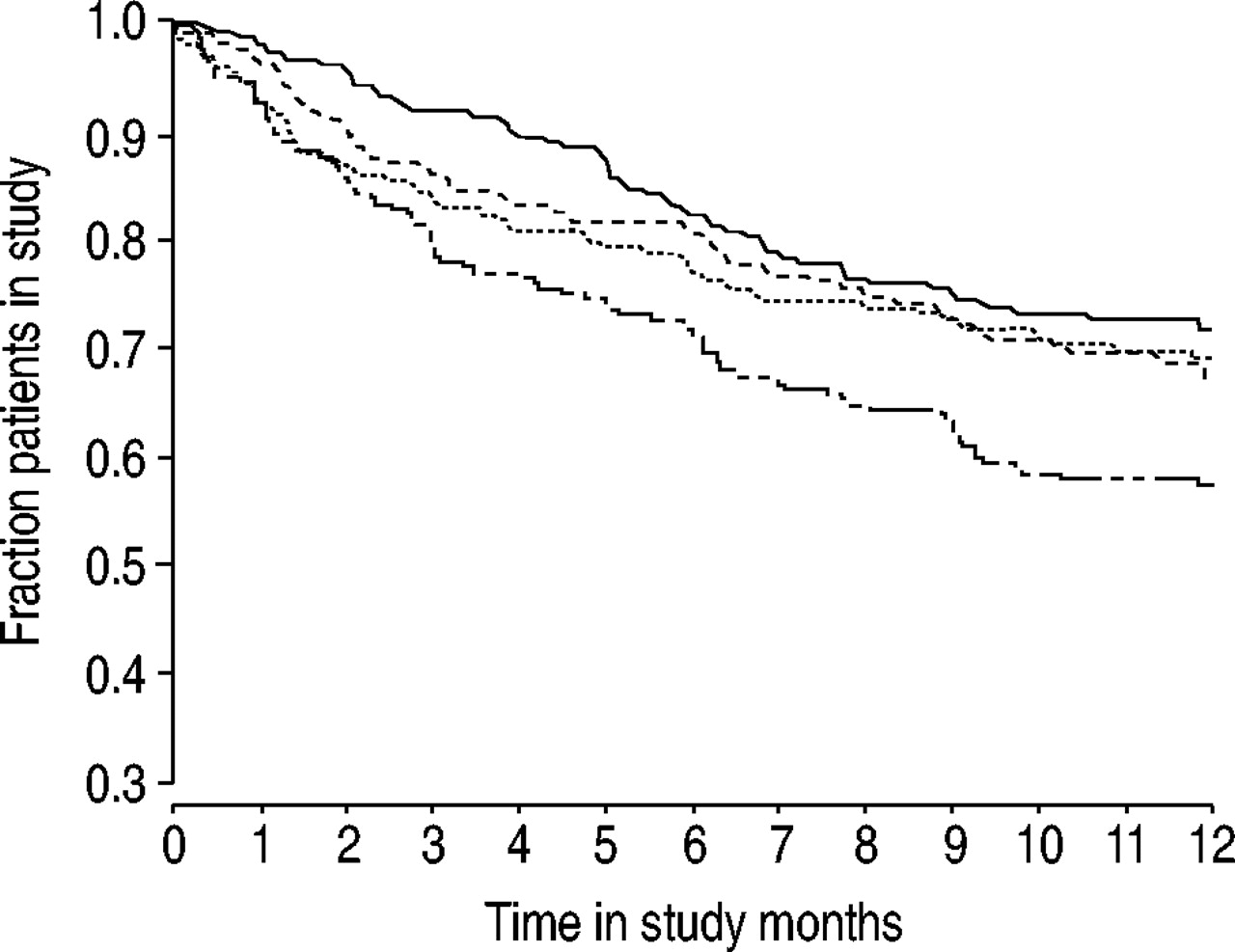
Kaplan-Meier plot of discontinuations, by treatment group. ––: Symbicort; ·····: budesonide; ----: formoterol; –··–: placebo.
Patient flow
Exacerbations
Severe
Mean exacerbation rates were 1.42, 1.59, 1.84 and 1.87 exacerbations per patient per year in the budesonide/formoterol, budesonide, formoterol and placebo treatment groups, respectively. Budesonide/formoterol significantly reduced the mean exacerbation rate versus placebo and formoterol (table 3⇓). Although not statistically significant, budesonide did produce a 15% reduction in severe exacerbations versus placebo. However, budesonide/formoterol reduced severe exacerbations to a greater extent than both budesonide and formoterol separately. There was no evidence of heterogeneity in the treatment differences in any of the treatment interaction analyses of exacerbation rates for sex, smoking status/smoking history, reversibility or use of inhaled corticosteroids at entry.
Statistical analysis of primary variables
The lowest rates of oral steroid courses associated with exacerbations were found in the budesonide/formoterol and budesonide groups (0.74 and 0.76 oral steroid courses per patient per year, respectively, compared with 1.04 for the formoterol and 1.07 for the placebo groups). Compared with placebo, both budesonide/formoterol and budesonide reduced the number of oral steroid courses used in association with exacerbations (31%, p=0.027 and 29%, p=0.045, respectively, compared with 3% for formoterol versus placebo, p=0.853). Budesonide/formoterol also significantly reduced the number of oral steroid courses compared with formoterol (28%, p=0.039).
Mild
All active treatment arms reduced mild exacerbations versus placebo: budesonide/formoterol by 62%, budesonide by 41% and formoterol by 55% (all p<0.001). Budesonide/formoterol reduced mild exacerbations by 35% versus budesonide (p=0.022) and by 15% versus formoterol (p=0.403).
Lung function
Forced expiratory volume in one second and vital capacity
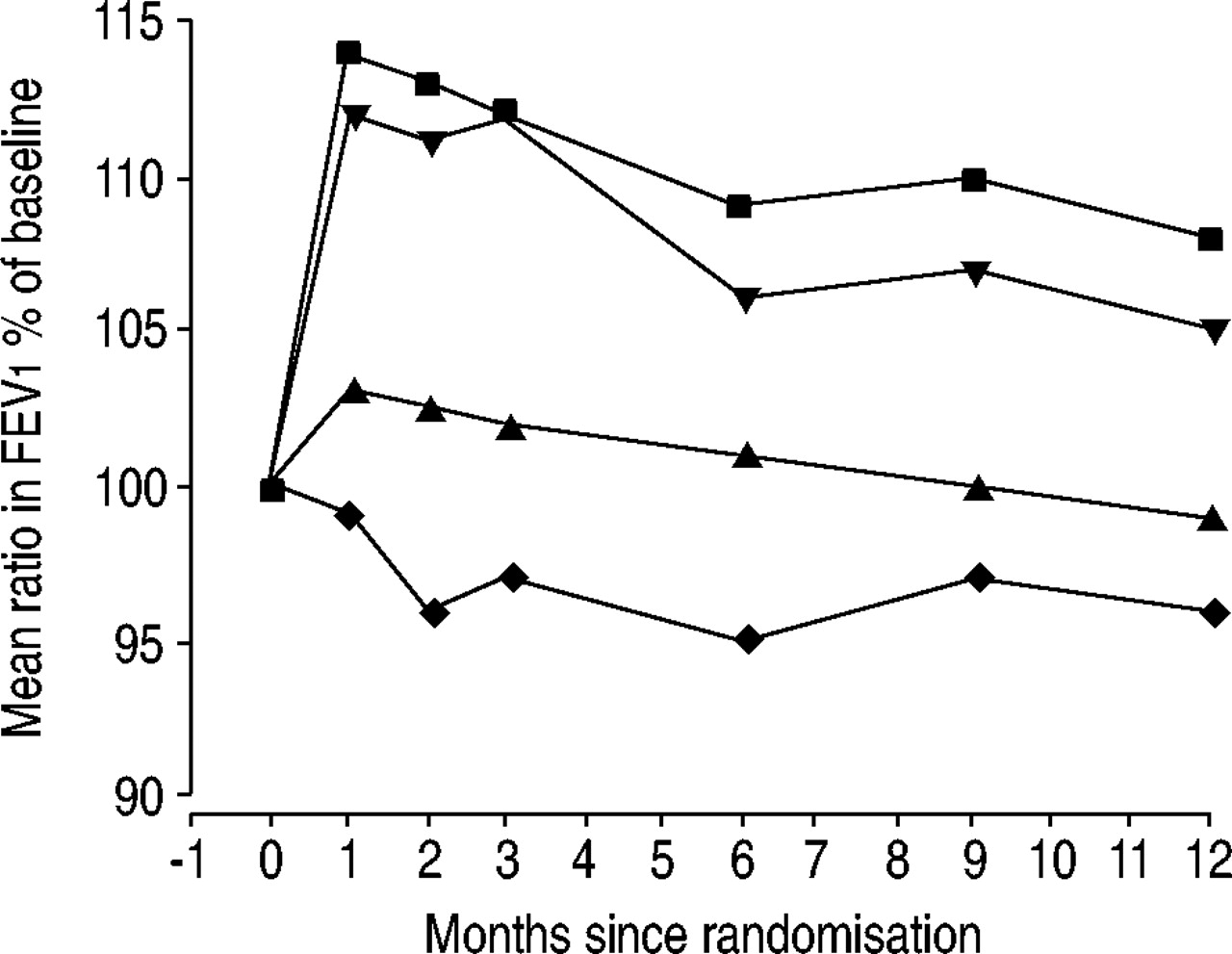
During the 12-month period, all active treatments increased FEV1 versus placebo; budesonide/formoterol also increased FEV1 versus budesonide (table 3⇑). The improvements in FEV1 were sustained with budesonide/formoterol throughout the study period compared with budesonide and placebo (fig. 2⇓). There was no evidence of heterogeneity in the treatment differences in any of the treatment interaction analyses of lung function for sex, smoking status/smoking history, reversibility or use of inhaled corticosteroids at entry. All active treatments improved VC compared with placebo (9%, p<0.001; 4%, p<0.05; 11%, p<0.001 for budesonide/formoterol, budesonide and formoterol, respectively).

Mean values for forced expiratory volume in one second (FEV1) measured at clinic visits. ▪: budesonide/formoterol; ▴: budesonide; ▾: formoterol; ♦: placebo. p<0.001 budesonide/formoterol versus placebo and budesonide; p<0.001 formoterol versus placebo; p<0.05 budesonide versus placebo.
Peak expiratory flow
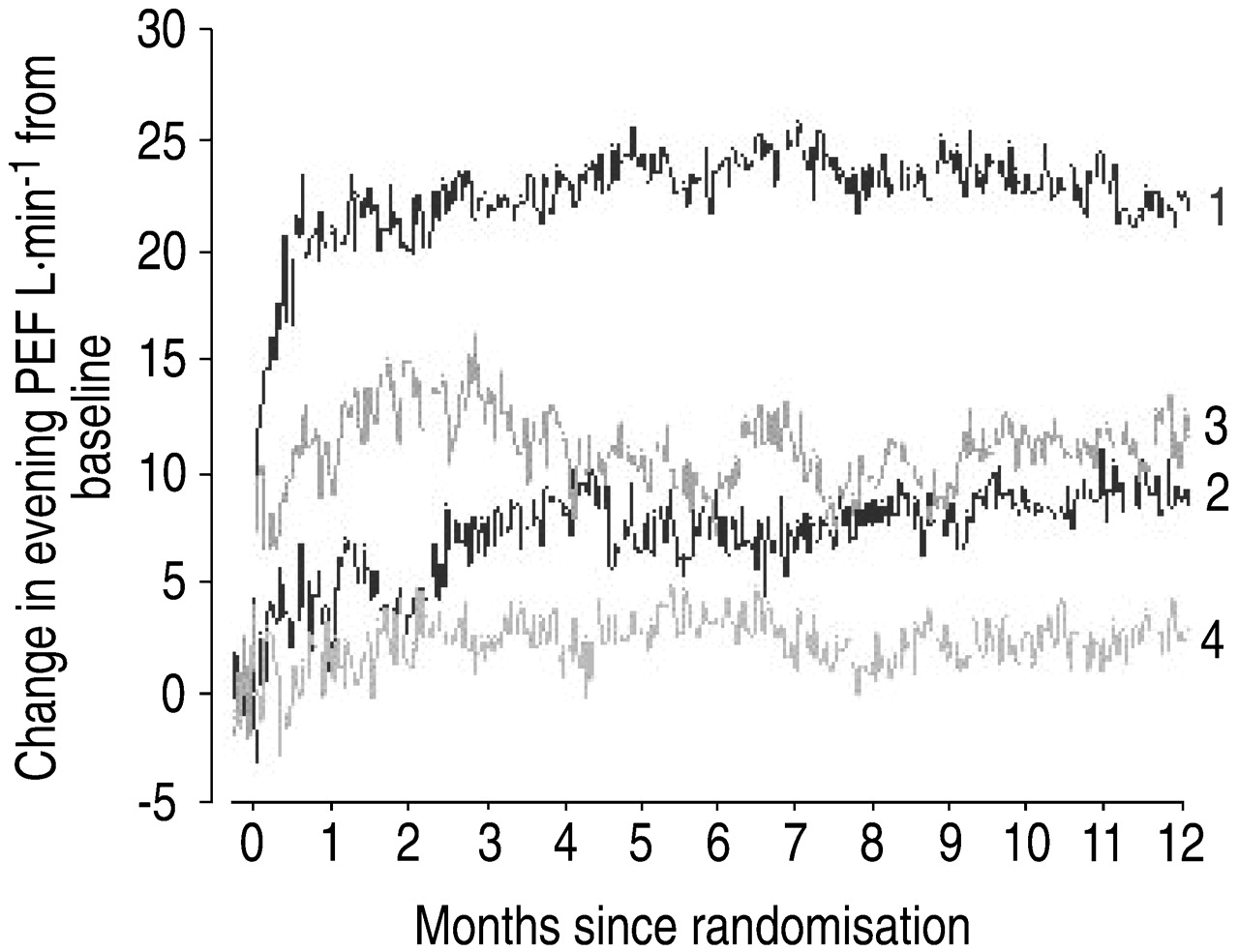
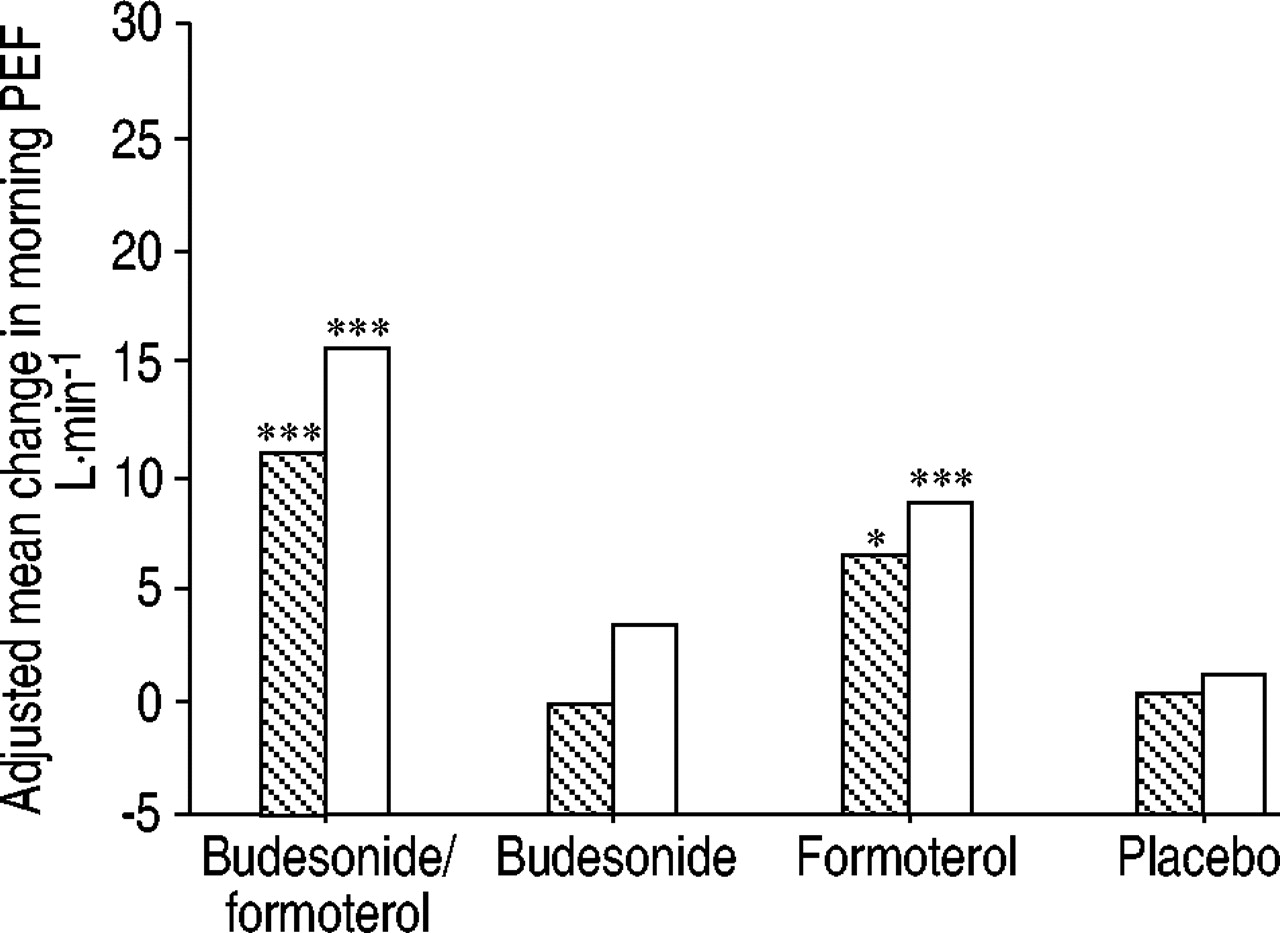
Budesonide/formoterol improved and maintained morning (fig. 3⇓) and evening (fig. 4⇓) PEF compared with placebo, budesonide and formoterol alone (all p<0.001). In a post-hoc analysis, budesonide/formoterol was shown to improve morning PEF as early as day 1 (fig. 5⇓). This improvement in morning PEF had further increased in the first week (fig. 5⇓) and was sustained at 12 months (fig. 3⇓).

Change in morning peak expiratory flow (PEF) from baseline over 12 months (from daily diary card data). 1: budesonide/formoterol; 2: budesonide; 3: formoterol; 4: placebo. Differences of 16, 12, 24 L·min−1 for budesonide/formoterol versus budesonide, formoterol and placebo, respectively (all p<0.001); budesonide versus placebo 8 L·min−1 (p=0.015); formoterol versus placebo 12 L·min−1 (p<0.001).

Change in evening peak expiratory flow (PEF) from baseline over 12 months (from daily diary card data). 1: budesonide/formoterol; 2: budesonide; 3: formoterol; 4: placebo. Difference of 15, 11, 20 L·min−1 for budesonide/formoterol versus budesonide, formoterol and placebo, respectively (all p<0.001); budesonide versus placebo 5 L·min−1 (p=0.104); formoterol versus placebo 9 L·min−1 (p=0.009).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Improvements in morning peak expiratory flow (PEF) from daily diary card data (day 1 (┘) and week 1 (□)). Day 1: budesonide/formoterol versus budesonide p<0.001, versus formoterol p=0.081. Week 1: budesonide/formoterol versus budesonide p<0.001, versus formoterol p=0.002. *: p<0.05 versus placebo; ***: p<0.001 versus placebo.
Symptoms
Budesonide/formoterol significantly reduced all symptom scores within the first week of treatment versus budesonide, formoterol and placebo; this significant effect was sustained for 12 months for budesonide/formoterol versus placebo, budesonide and, for formoterol, regarding the total score and awakenings (table 4⇓). Budesonide/formoterol increased days free from shortness of breath by 12% versus placebo (p<0.001) and awakening-free nights by 14% versus placebo (p<0.001), each equivalent to approximately one extra day/night per week. Budesonide/formoterol reduced use of reliever medication by 1.3 and 0.7 inhalations per 24 h versus placebo and budesonide, respectively (both p<0.001).
Symptom scores for first week of treatment and at 12 months
Health-related quality of life
Baseline values for SGRQ total score were between 51–54 in the four groups. Mean reductions from baseline were −3.9, −1.9, −3.6 and −0.03 after budesonide/formoterol, budesonide, formoterol and placebo treatment, respectively. A change of 4 points from baseline is considered as an important difference relevant to the patient. Compared with placebo, budesonide/formoterol significantly improved SGRQ total score (mean difference 3.9, p=0.009), and symptom (mean difference 5.9, p<0.001) and impact (mean difference 4.7, p=0.006) domains.
Safety
The AE profile was similar in each group, although proportionally more patients reported COPD events in the placebo group than in the active treatment groups (17, 13, 19 and 26% in the budesonide/formoterol, budesonide, formoterol and placebo groups, respectively). No treatment-related patterns were discernable regarding the incidence or cause of death (n=26) or serious AEs (table 5⇓). Discontinuations due to COPD worsening were highest in the placebo group (table 2⇑). The frequency of discontinuations due to other AEs was similar in all groups (table 2⇑). No clinically-important between-group differences were identified for laboratory (clinical chemistry and haematology) or ECG measurements (including Q‐T interval corrected for heart rate prolongation) and there were no reported cases of bruising.
Serious adverse events (SAEs)
Discussion
This study showed that 1‐yr treatment with budesonide/formoterol in a single inhaler resulted in significant improvements versus placebo in all important clinical disease parameters characterising COPD. The results support the authors' hypothesis that the effect of budesonide/formoterol on the primary efficacy variables of severe exacerbations and lung function was mainly attributable to the steroid component and to the β2‐agonist component, respectively.
One of the most important benefits of budesonide/formoterol treatment can be seen as the reduction in the mean number of severe exacerbations. This result is probably an underestimation rather than an overestimation of the effect of budesonide/formoterol, especially versus placebo, because of the greater withdrawal rate in the placebo group; it can be assumed that it was the most severely ill patients who dropped out, potentially resulting in fewer exacerbations in the placebo group. Considering that several weeks of convalescence may be needed to recover from a single exacerbation, perhaps 6–8 weeks for full recovery 26, the reduction in exacerbation rate with budesonide/formoterol could be considered clinically and economically important. Using these figures of 6–8 weeks, it can be seen that a COPD patient experiencing, for example, two exacerbations per year may be effectively disabled for up to 16 weeks during this time; a reduction in the number of exacerbations of 24%, as shown for budesonide/formoterol versus placebo in this study, could represent a meaningful benefit of perhaps 4 weeks less disability per year for the patient together with associated cost savings to society. In addition, the requirement for less oral steroid associated with exacerbations by patients taking budesonide/formoterol will contribute to reducing the systemic steroid load of these patients.
Improvements in lung function are in line with results of previous bronchodilator studies with formoterol in COPD 10, 27, 28 and were sustained throughout the study period compared with comparators. In general, a mean increase of 15% in FEV1, as found here for budesonide/formoterol versus placebo, can be considered as clinically relevant in COPD. This is supported by a study which found that a mean increase of 114 mL in patients with FEV1 of 35% pred was associated with a noticeable difference in dyspnoea 29. Of particular interest, is that patients in the current study were suffering from more severe COPD in terms of spirometry than patients in the study by Burge et al. 18. These authors observed improvements with high doses of inhaled fluticasone, yet the patients in the present study gained similar benefits with modest doses of budesonide singly and in combination. Indeed, this is the first data to show that conventional doses of budesonide, particularly in combination, do work in this severity of disease, producing benefits in terms of lung function, exacerbations and health status.
An important finding was the early improvement in morning PEF in the budesonide/formoterol group, where a significant increase was observed in the first morning after starting treatment. This increase in morning PEF was further accentuated after 1 week's treatment compared with placebo, budesonide alone and formoterol alone and maintained throughout the study period. Rapid onset of bronchodilation is probably important both when starting and during treatment in providing immediate relief, potentially increasing confidence in the drug and aiding adherence to the treatment.
Interestingly, the improvements in morning PEF as well as evening PEF were significantly greater in the budesonide/formoterol group compared with formoterol alone, which implies a contribution by the inhaled steroid and is probably linked to the improvement seen in symptoms. This obvious additive effect emphasises the potential advantage of using budesonide/formoterol in a single inhaler in COPD. Furthermore, these results seem to indicate that daily PEF is a useful tool for consistent longitudinal measurement to discriminate between treatments when evaluating drug effects in COPD. This would suggest a greater role for PEF in COPD than has been previously accepted.
In addition to reducing the rate of severe exacerbations and improving lung function, budesonide/formoterol improved all COPD symptoms (breathlessness, chest tightness, cough and night-time awakenings) according to the symptom scores. This improvement in symptoms versus placebo provided approximately one extra day/night per week free from shortness of breath and night-time awakenings, respectively. It seems likely that these improvements were clinically relevant and important. This judgement is supported by the fact that there was also a significant decrease in the total score of the SGRQ in the budesonide/formoterol group compared with placebo, thus reflecting that budesonide/formoterol treatment resulted in a better HRQL. A significant reduction in the use of reliever β2‐agonists was documented with budesonide/formoterol, together with a reduction in the rate of mild exacerbations. This further supports the clinical benefits of budesonide/formoterol in COPD.
During this 12-month study a large number of patients (34%) discontinued. Most discontinuations were seen in the placebo group, where more than twice as many patients than in the budesonide/formoterol group discontinued because of deteriorating COPD. This difference was statistically significant, indicating possible treatment failures. The budesonide/formoterol group had the largest number of patients completing the study. Potentially, the differences in discontinuations could lead to bias in treatment estimates, usually favouring treatments with higher withdrawal rates e.g. placebo, since data used in the analysis will be from a “nondeteriorating” period. Thus, results in this study may be conservative (underestimated) as compared with placebo.
Twenty-six patients died during the study year, which is not unexpected in a COPD population of moderate-to-severe disease. These cases were uniformly distributed between the groups. All treatments were well tolerated and the side-effect profiles were similar in the four treatment groups. Earlier studies with budesonide and formoterol support the long-term safety of budesonide/formoterol in the treatment of patients with COPD. The 3‐yr European Respiratory Society study on COPD (EUROSCOP), for example, showed no difference in change in bone mineral density with budesonide 800 µg·day−1 or with placebo 30, 31, while two 12-month trials with formoterol versus placebo showed no significant differences between treatments in the proportion of patients reporting cardiovascular AEs 32.
The effect of budesonide/formoterol on the rate of severe exacerbations is clinically most important and agrees with the results seen with this treatment in patients with persistent asthma 33, 34. The complementary therapeutic interactions between the anti-inflammatory corticosteroid budesonide and the rapid- and long-acting β2‐agonist formoterol may explain why the combination is clinically useful in both diseases. The use of one single inhaler simplifies treatment, should improve adherence, and increases the likelihood that patients obtain the corticosteroid component of the medication.
Conclusions
Budesonide/formoterol was shown to be an effective treatment for the management of moderate-to-severe chronic obstructive pulmonary disease, reducing exacerbations and providing early and sustained improvements in lung function and symptoms, together with improvements in health-related quality of life. Budesonide/formoterol demonstrated a similar safety profile to placebo. Budesonide/formoterol provided clinically meaningful benefits to chronic obstructive pulmonary disease patients and, therefore, has the potential for reducing the burden of the disease. These results suggest a role for budesonide/formoterol in the long-term management of moderate-to-severe chronic obstructive pulmonary disease.
Acknowledgments
The authors would like to acknowledge the contribution of R. Pauwels in the study design, thank E. Bateman for discussions on the manuscript and thank all of the investigators who recruited and treated patients.
- Received April 16, 2002.
- Accepted August 5, 2002.
- © ERS Journals Ltd
References









