





Abstract
Overdose of tricyclic antidepressants, which inhibit cellular serotonin (5-HT) uptake, sometimes causes acute respiratory syndrome-like symptoms. Their acute and chronic cardiopulmonary actions, which might be implicated, utilising both in vivo and ex vivo animal studies, were investigated in this study.
Acute amitriptyline (AMI), iprindole and imipramine caused dose-dependent prolonged rises in pulmonary artery pressure and oedema in anaesthetised cats in vivo. Acute AMI, in isolated ex vivo blood-perfused rat lungs, also caused dose-dependent sustained vasoconstriction, which could be attenuated with either calcium channel inhibition or a nitric oxide donor. It was demonstrated that the pressor effects of AMI were not due to release of histamine, serotonin, noradrenaline, or the activities of cycloxygenase or lipoxygenase. After AMI, hypoxic pulmonary vasoconstriction and the pressor actions of 5‐HT and noradrenaline were diminished, possibly due to uptake inhibition. Activities of the endothelial-based enzymes, nitric oxide synthase and endothelin-converting enzyme, were undiminished. Large acute doses of AMI caused oedema with rupture of capillaries and alveolar epithelium.
Chronic iprindole raised pulmonary artery pressure and right ventricle (RV)/left ventricle (LV) + septal (S) weight. Chronic AMI led to attenuation of the pressor action of 5‐HT, especially when associated with chronic hypoxic-induced pulmonary hypertension. RV/LV+S weight increased, attributable to LV decline.
The acute and chronic effects observed might have relevance to clinical overdose, while the attenuation of acute effects offers possible therapeutic options.
This work was partly supported by a grant from the British Lung Foundation.
The tricyclic antidepressant (TCA) drugs, amitriptyline (AMI), iprindole and imipramine, are amphiphilic compounds with a hydrophobic ring structure and a primary or substituted amine side-chain bearing a net-positive charge. Their amphiphilic nature causes them to interact with phospholipids, producing generalised phospholipidosis when given chronically to rats and mice; foam cells were seen in the alveoli after chronic treatment with iprindole and imipramine but not with AMI 1. These drugs affect serotonin (5‐HT) uptake in cells, especially platelets. A high tissue/blood ratio has been reported for AMI, with the lung showing a high affinity for the drug 2. TCAs show complex interactions with vasoactive autacoids. They are anticholinergic, prevent breakdown of noradrenaline (NA) and may inhibit synthesis of prostaglandins.
TCAs have chemical similarities with certain anorectic drugs, which also affect 5‐HT transport and have been implicated as risk factors for primary pulmonary hypertension. In a parallel study, the current authors found that the anorectics fenfluramine and chlorphentermine have pulmonary vascular effects and influence certain endothelial functions 3.
TCAs are frequently used in self poisoning, particularly AMI. In the early stages, death commonly results from cardiac arrhythmias, but there are now many records of delayed death due to lung complications and radiography often shows lung damage in the early stages. However, there is no record of long-term pulmonary problems following prolonged treatment at normal dose levels.
The aim of this study was to establish the cardiopulmonary effects of TCAs which might underlie the clinical consequences of overdose. The study was conducted at two centres (University of Sheffield, Sheffield, UK and 2nd Medical School, Prague, Czech Republic). The acute pulmonary vascular actions of overdose levels of the antidepressants AMI, imipramine and iprindole, as well as the consequences of chronic treatment with AMI and iprindole, were examined.
Methods
Ethical considerations
This was a two-centre study. All procedures were performed in strict accordance with the regulations of the appropriate authorities.
Animals
Cats (1.4–5.1 kg) were anaesthetised with chloralose (100 mg·kg−1 i.p.) and rats (Wistar strain, 200–350 g) with pentobarbitone (60 mg·kg−1 i.p.). Heparin was given intravenously (1000 units·kg−1).
Cat left lower lobe preparation
In anaesthetised, open-chest cats (ventilated with a Starling pump; Havard Instruments, Kent, UK), the left lower lobe of lung was perfused in vivo at a constant flow with autologous blood from the cannulated right atrium, as described previously 4; thus, changes in pulmonary artery pressure (Ppa) represented changes in pulmonary vascular resistance. Lobar flow was maintained at 100 mL·kg−1 (∼1/6th cardiac output), which resulted in pressure measurements within the normal range. The lobe was separately ventilated with a second Starling pump through the cannulated bronchus, and bronchial pressure was measured (Pbr). Left atrial pressure (Pla) was measured from a cannula in the atrial appendage and systemic blood pressure (Psys) from a femoral cannula. TCAs were given i.v. in increasing doses; effects were prolonged, but further doses were delayed until initial conditions were restored. Saline control injections were given in all experiments and were without effect.
Isolated ex vivo blood-perfused rat lungs
After anaesthesia, rat lungs in situ were perfused with homologous blood at a constant flow (20 mL·min−1) at 38°C, as described previously 5. After heparinisation the chest was opened with the lungs left in situ; the pulmonary artery and left atrium were cannulated and blood was circulated from a heated reservoir. Ppa was measured close to the cannulated pulmonary artery. The lungs were ventilated with air+5% carbon dioxide (CO2) (normoxia). Drugs were injected into the circuit close to the pulmonary artery and saline injections of equal volume were given as controls. Ventilation of the lung with an hypoxic gas (2% oxygen (O2)+5% CO2) caused a stable rise in Ppa (hypoxic pulmonary vasoconstriction (HPV)).
Electron microscopy
AMI (Roche, Hertfordshire, UK) was added into the circuit of the perfused rat lung. Ppa was monitored and the physiological effect was recorded. The lungs were then removed and perfused via the pulmonary artery, with glutaraldehyde at 20 mmHg while the lung was inflated to 20 cmH2O, and processed for electron microscopy.
Chronic amitriptyline and iprindole treatment
Male litter mates were divided into treated and untreated groups.
Experiment 1: amitriptyline i.p. in normoxic rats
Eight rats received 25 mg·kg·day−1 AMI i.p. and seven controls received equivolume isotonic saline (0.9% NaCl), for 15 days. After anaesthesia the Ppa was measured by cardiac catheterisation via the right jugular vein in the close-chested rat 6. The heart was then removed and the right ventricle (RV)/left ventricle (LV)+septum (S) were weighed.
Experiment 2: amitryptyline i.p. in normoxic and chronically hypoxic rats
Groups of six rats were held in normoxia or chronic hypoxia in a normobaric environmental chamber for 21 days. The chamber, described previously 5, maintained O2 at 10% and CO2 stable at ∼0.2%. Litters of six rats were split, three were put into the chamber and three were kept in the same room in air, three rats per cage. AMI, 25 mg·kg−1, was given i.p. daily for 21 days and controls received saline. After treatment the isolated blood-perfused lung preparation was set up as described above. Pulmonary vascular resistance was calculated as the slope of the pressure/flow relationship. Briefly, the flow rate was reduced in a stepwise fashion (20, 15, 10, 5, 0 mL·min−1) and the Ppa was allowed to stabilise. The extrapolated intercept on the pressure axis, derived from the linear portion of the relationship (5–20 mL·min−1), gave an indication of critical closure pressure. The pulmonary vascular response to 5‐HT (25, 50 µg; Sigma, Poole, UK) and angiotensin I (AI, 0.5 µg; Sigma) was also recorded. Saline injections of a similar volume were given and were without effect.
Experiment 3: amitryptyline per os normoxic and chronically hypoxic rats
There were five rats in both the normoxic and chronic hypoxic groups. AMI, as a tryptazol paediatric syrup (25 mg·kg·day−1; Royal Hallamshire Hospital, Sheffield, UK), was given by gavage for 21 days and controls received syrup alone. After anaesthesia, the lungs were fixed with 10% buffered formalin via the trachea at an inflation pressure of 20 cmH2O for histological analysis, and RV/LV+S was measured as in Experiment 1.
Experiment 4: iprindole i.p. in normoxic rats
In Experiment 4a, five rats received iprindole (25 mg·kg·day−1 i.p.; Wyeth Laboratories, Hampshire, UK) and six rats received saline for 15 days. Ppa and RV/LV+S were measured as in Experiment 1.
In Experiment 4b, 10 rats received iprindole (25 mg·kg·day−1 i.p.) and nine rats saline for 15 days. RV/LV+S was measured as in Experiment 1.
Light microscopy
After wax embedding, 5 µm-thick sections were stained with Gomori's elastic stain to visualise the elastic laminae in the vascular wall. The development of a double elastic lamina enclosing new medial muscle is indicative of vascular remodelling associated with pulmonary hypertension. The percentage of small (≤50 µm diameter) pulmonary arterioles, adjacent to alveoli, with a double elastic laminae around ≥50% of the vessel circumference (% thick walled pulmonary vessels (%TWPV) 7) was ascertained under light microscopy (magnification ×400). This method is capable of detecting small changes not detected by medial thickness measurements.
Statistics
Means and sem were calculated and compared by paired and unpaired t‐tests as appropriate. %TWPV was assessed by the Mann-Whitney U‐test. Differences were considered significant when p<0.05.
Results
Acute studies
Cat left lower lobe preparation
AMI (n=8, 0.5–10 mg), iprindole (n=6, 1–10 mg) and imipramine (n=3, 1.25–12.5 mg), given into the inflow tubing to the lobar circuit, gave prolonged dose-dependent increases in Ppa and bronchoconstriction, and oedema often followed. Figure 1a⇓ shows superimposed traces of Ppa changes after 0.5–10 mg AMI. Similar results followed treatment with iprindole and imipramine. Figure 1b⇓ shows the effect of 5 mg iprindole on Ppa, Pbr, Psys, Pla and flow to the lobe (constant). All three drugs led to changes in Psys and Pbr and occasional changes in Pla, which were insufficient to contribute to the large changes in Ppa. Similar volumes of saline were given in all experiments and had no pressor effect.

Traces of pulmonary artery pressure (Ppa) in a cat left lower lobe preparation with a constant blood flow. a) Successive doses of amitriptyline (AMI; 0.5 (—), 1 (– - –), 2 (- - -), 5 (—) and 10 mg (····); arrow indicates administration of drug) are superimposed. Ppa remains raised between doses. b) Iprindole causes a large persistent rise in Ppa (- - -), a transient fall in systemic blood pressure (Psys; ····), a prolonged rise in bronchial airway pressure (Pbr; double headed arrow), but negligible changes in left atrial pressure (Pla; – - –).
Rat isolated ex vivo blood-perfused lung
In seven rats, dose-response relationships were established using increasing doses of AMI (0.1, 0.2, 0.3, 0.4, 0.5, 1, 2 and 5 mg) injected into the inflow tubing of the circuit (cumulative perfusate concentration 3×10−5–2×10−3 M). They caused brief falls in Ppa, followed by persistent increases with doses ≥0.2 mg (figs. 2⇓ and 3⇓). After large doses, further doses led to falls in Ppa and oedema often supervened. Oedema was not directly due to high Ppa as it also occurred when baseline Ppa was low (fig. 4⇓). Similar volumes of saline had no pressor effect.
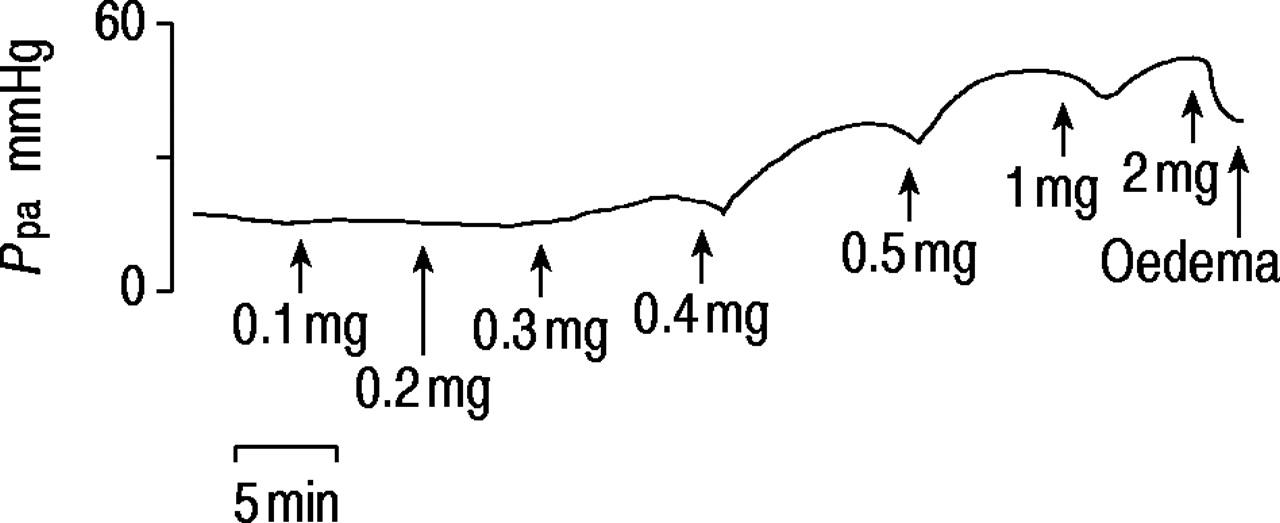
Trace of pulmonary artery pressure (Ppa) in isolated perfused rat lung showing responses to successive doses of amitriptyline. The effects are cumulative. The final dose caused dilatation and oedema.

Mean cumulative rises in pulmonary artery pressure (Ppa) after 0.6, l, 1.5 and 2.5 mg amitriptyline (AMI) in seven rats.

Trace of pulmonary artery pressure (Ppa) in an isolated rat lung showing abolition of the pressor response to serotonin (5‐HT) by ketanserin. Subsequently, there is a large rise in Ppa after 0.5 mg amitriptyline (AMI) and a huge rise, leading to oedema, after 5 mg AMI.
Tests for endothelial function
Angiotensin converting enzyme activity
Angiotensin converting enzyme (ACE) activity was tested with angiotensin I (AI, n=4) and bradykinin (BK, n=4). Neither activity was altered after AMI (0.5 mg). AI (0.5 µg, repeated twice) caused rises in Ppa of 5.8±0.9 before and 7.4±1.6 mmHg after AMI (ns). BK (10 µg, twice) caused rises in Ppa of 8.7±1.1 before and 8.3±1.1 mmHg after AMI (ns).
Endothelin converting enzyme and nitric oxide synthase activity
There was no evidence that either endothelin converting enzyme or nitric oxide synthase (NOS) activity was reduced after AMI. Big endothelin‐1 (1 μg; Sigma) caused a similar rise in Ppa either in the presence (n=5) or absence (n=5) of 0.5 mg AMI (ΔPpa 5.7±2.0 mmHg versus 2.8±0.4 mmHg, ns). The rise in Ppa with 100 µg (1×10−5 M) N‐nitro‐L‐arginine methyl ester (L‐NAME) in the presence of AMI (fig. 5a⇓) (ΔPpa 5.2±1.6 mmHg) suggests that AMI stimulated NOS activity, because L‐NAME does not usually cause substantial rises in Ppa in normal rat lungs during normoxia 8.

a) Trace of pulmonary artery pressure (Ppa) in a rat lung showing that the pressor effect of hypoxia (2% oxygen (O2)+5% carbon dioxide (CO2)) is greatly reduced after amitriptyline (AMI). In this rat, AMI caused dilatation only. After AMI, N‐nitro‐L‐arginine methyl ester (L‐NAME, 100 µg, 1×10−5 M) caused a rise in Ppa. Subsequently, the pressor response to hypoxia was greatly increased. b) Trace of Ppa in a rat lung showing persistent large rises in Ppa after noradrenaline (NA), followed by a test with hypoxia. After AMI, which caused only a small rise in Ppa, the effect of hypoxia was reduced and the response to NA was negligible.
Pressor effects of amines
5‐HT (100 µg), given twice with reproducible results, caused a rise in Ppa of 8.9±1.9 mmHg before but a significantly smaller rise of 1.7+0.05 mmHg after 0.5 mg AMI (n=6, p<0.05). Similarly, NA (100 µg) was given twice before and twice after 0.5 mg AMI. The pressor response was reduced (ΔPpa 1.0±0.8 mmHg before, 0.06±0.05 mmHg after AMI p<0.05, n=4, fig. 5b⇑).
Effect on hypoxic pulmonary vasoconstriction
A reproducible HPV (rise in Ppa) was established after 2–3 hypoxic challenges (fig. 5a⇑). Subsequently, 0.5 mg AMI, which caused a persistent stable rise in Ppa, reduced HPV. With hypoxia, Ppa rose 7.3±1.8 mmHg before and 2.7±0.7 mmHg after AMI (p<0.05). After the addition of 100 μg (1×10−5 M) L‐NAME, HPV was restored (fig 5a⇑). Unusually, AMI caused only a small dilatation and no constriction.
Tests for secondary release of autacoids by amitryptyline
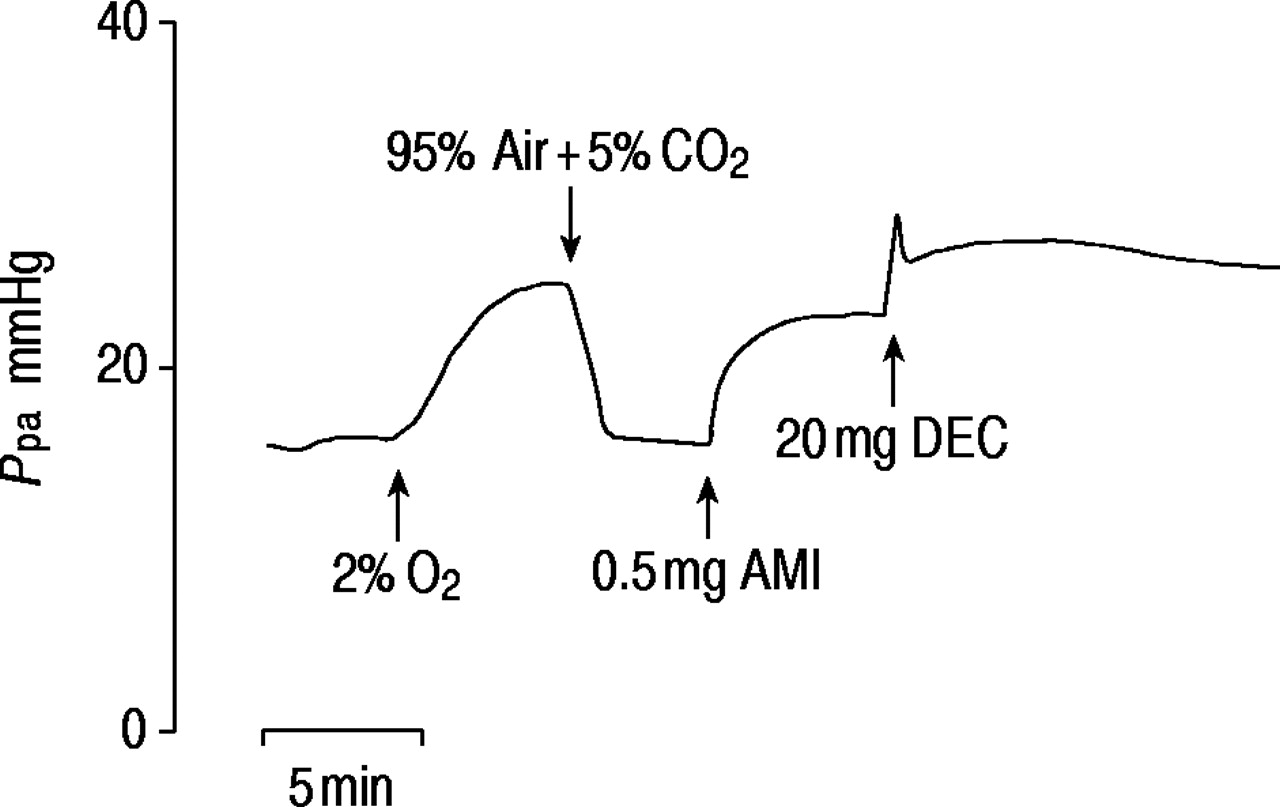
The pressor/dilator action of AMI could not be inhibited with inhibitors of potential mediator receptor/enzymes (table 1⇓). These included: cyclooxygenase with meclofenamate (100 µg, 1×10−5 M), which blocked the pulmonary vasomotor effects of arachidonic acid 9; lipoxygenase with diethylcarbamazine (DEC) (20 mg, 5×10 M), which abolished HPV; α‐adrenoreceptors with phentolamine (0.2 mg, 1×10−5 M), which abolished the pressor effect of NA; and 5‐HT receptors with ketanserin (1×10−5 M), which abolished the pressor effect of 5‐HT (fig. 4⇑). Histamine H1 and H2 blockade was achieved with piriton (1 mg, 3×10−4 M) and cimetidine (2.5 mg, 8×10−4 M) in doses found to be effective in previous studies 10. Due to persistence of response, AMI was only given after the addition of the inhibitor into the blood perfusate. Table 1⇓ shows that the increases in Ppa with AMI were within the normal range and that oedema still followed large doses. DEC (20 mg, 5×10−4 M), given after the rise in Ppa caused by AMI, did not reduce Ppa but caused a further rise (2.0±0.7 mmHg, n=4) (fig. 6⇓).

Trace of the effect of diethylcarbamazine (DEC) after amitriptyline (AMI). Ppa: pulmonary artery pressure; O2: oxygen; CO2: carbon dioxide.
Effect of mediator receptor/enzyme blockade on the pressor response to amitriptyline (AMI) in isolated blood perfused rat lungs
Effect of dilator drugs after amitryptyline
The effect of s‐Nitroso‐N‐acetylpenicillamine (SNAP, NO donor; Tocris Pharmaceuticals, Avonmouth, UK) and verapamil (Ca2+ channel inhibitor; Abbott Laboratories, IL, USA) on Ppa, after Ppa was raised with 0.5 mg AMI, was tested (fig. 7⇓). SNAP (100 µg, 5×10−5 M, n=4) caused a large reduction in Ppa after AMI, −9.8±1.8 mmHg. Verapamil (100 µg, 1×10−5 M, n=7) also reduced Ppa after AMI, −3.2±0.4 mmHg.

Trace of the dilator action of s‐Nitroso‐N‐acetylpenicillamine (SNAP) (5×10−5 M), after pulmonary artery pressure (Ppa) was raised by amitriptyline (AMI). O2: oxygen; CO2: carbon dioxide.
Electron microscopy after acute amitryptyline
Electron micrographs, taken from rat lungs treated acutely with medium (200 µg) and large (4 mg) doses of AMI, showed damage to the alveolar lining and alveolar capillary endothelium (fig. 8⇓). There was distension and rupture of capillaries and oedema and red blood cells in the alveolar space; alveolar lining cells were displaced by oedema and formed bullae projecting into the alveoli.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electron micrographs after amitriptyline (AMI) administration to isolated rat lungs. After 4 mg AMI (a), the capillaries in the alveolar walls have dilated and have ruptured (arrows); fluid and red blood cells are present in alveolar space. After 200 µg AMI (b and c), type 1 cells have detached from the basal lamina and blebs protrude into the alveolar space.
Chronic studies
Chronic amitriptyline
Experiment 1 (table 2⇓). AMI-treated rats had a tendency to higher Ppa, but the difference was not significant. RV/LV+S was significantly raised in the AMI group. Absolute RV weight was unchanged but LV+S weights were reduced and account for the increase in the RV/LV+S ratio. The treated rats were in poor condition, which may have affected cardiac output and Ppa.
Effect of chronic amitriptyline treatment on indices of pulmonary hypertension in the rat
Experiment 2 (table 3⇓). AMI treatment had no effect on indices of pulmonary hypertension, i.e. slope of the pressure/flow (P/Q) line or intercept. Chronic hypoxic rats had steeper P/Q lines, higher intercepts on the Ppa axis and increased pressor responses to AI, as previously reported 5. The only difference in vasoreactivity observed between AMI and saline-treated rats was that chronic hypoxic AMI-treated rats had significantly reduced responses to 25 and 50 µg 5‐HT; the saline-treated chronic hypoxic rats had greater pressor responses to 5‐HT than normoxic rats. AMI-treated normoxic rats also had reduced responses to 5‐HT compared with saline-treated controls (ns).
Effect of chronic amitriptyline on pulmonary vascular resistance and response to vasoconstrictors (Experiment 2) and indices of pulmonary hypertension (Experiment 3) in ex vivo isolated blood-perfused rat lungs
Experiment 3 (table 3⇑). The %TWPV was not significantly altered by AMI treatment. In normoxic groups, unlike Experiment 1, neither absolute RV or LV+S weights or RV/LV+S ratio were significantly altered by AMI treatment. However, there was evidence of a significant reduction in LV+S/ body weight (BW) in the treated group (LV+S/100 g BW 0.22±0.002 and 0.21±0.003 untreated versus treated, respectively p<0.001). These rats were in better condition than those given AMI i.p. (Experiment 2).
Chronic hypoxia caused RV hypertrophy and an increase in RV/LV+S with no change in LV+S/BW, and an increase in %TWPV. There was no difference between AMI treated and untreated groups.
Chronic iprindole
Experiment 4a (table 4⇓). Ppa and RV/LV+S were raised after chronic iprindole treatment and BW was reduced (p<0.01).
Effect of chronic iprindole treatment on indices of pulmonary hypertension in the rat
Experiment 4b (table 4⇑). RV/LV+S was raised after treatment although not quite to significance (p=0.053), BW was reduced (p<0.001). Although the absolute LV+S was significantly reduced (p<0.001), when LV was expressed per BW there was no difference between treated and untreated groups (LV+S/100 g BW, control 0.23±0.003 versus iprindole 0.22±0.006, ns).
Discussion
Main findings
In this study, acute administration of overdose levels of the TCAs AMI, imipramine and iprindole caused persistent rises in Ppa in cats in vivo. This pressor effect of AMI, studied in ex vivo blood-perfused isolated rat lungs, was not due to release of histamine, NA, 5‐HT, products of cyclooxygenase or lipoxygenase but could be reduced by calcium channel inhibition or an nitric oxide (NO) donor. Chronic AMI reduced pulmonary vascular responses to the biogenic amines 5‐HT and NA. Although this may be due to a direct effect on amine receptors responsible for vasoconstriction, another possible mechanism is the inhibition of amine uptake by pulmonary endothelium. Chronic treatment with iprindole, but not AMI, led to pulmonary hypertension.
Overdose of tricyclic antidepressants
Self-poisoning by TCAs is common and increasing. Although cardiac complications are the common cause of death, some delayed fatal pulmonary complications were reported in the 1970s 11, 12. The lung accumulates these drugs 2, 13 and more recent large surveys have shown that lung radiological changes are frequent and may be an early development. Varnell et al. 14 reviewed 81 cases of TCA overdose (20 cases with AMI). Fifty-four per cent had radiological abnormalities in the lung while 9% had clinical and radiological changes consistent with acute respiratory distress syndrome (ARDS). Aspiration could account for some effects. Roy et al. 15 surveyed 82 consecutive patients with TCA overdose. The ratio of arterial to alveolar oxygen pressure was reduced and mechanical ventilation was required in 77%. Nearly half showed lung radiological abnormalities within the first 48 h. In 56 consecutive cases, Shannon et al. 16 also found a high frequency (30%) of abnormal lung radiography. It is therefore emerging that understanding the pulmonary changes and looking for suitable treatment is of great importance.
Acute administration of amitriptyline
Several of the acute effects observed with AMI in both cats in vivo and ex vivo isolated rat lungs may be relevant to the clinical problems seen after overdose 17. Persistent raised Ppa, oedema and suppression of hypoxic vasoconstriction was found. The latter could have serious consequences through impairment of ventilation/perfusion ratio matching when the lung is patchily affected. Depression of HPV may have been due to increased activity of endothelial NOS. It would be valuable to find out whether inducible NOS, which may be responsible for some aspects of ARDS, becomes expressed in these conditions. Evidence that AMI impaired the pressor action of both 5‐HT and NA in rat lungs was found. TCA drugs inhibit uptake of 5‐HT and NA, although this is through separate transporter mechanisms 18. Systemic consequences of impaired amine clearance by the lung in TCA overdose could be important. It is known that catecholamine blood levels are raised in TCA overdose; this in turn might lead to the lipid disturbances which have been observed.
The electron micrographs in this study showed that one large acute dose of AMI could cause rupture of the alveolar lining, breaks in capillary endothelium and fulminant oedema. However, a smaller dose, although sufficient to cause some ultrastructural damage, did not impair endothelial enzyme activity (endothelial NOS, cyclooxygenase and endothelin converting enzyme).
The rise in Ppa with acute doses of AMI could be reduced substantially by addition of either a calcium channel inhibitor or an NO donor, suggesting that these changes are reversible and thus treatable.
Chronic treatment with amitriptyline
Impaired 5‐HT pressor responses persisted after chronic treatment with AMI. The rise in Ppa after 5‐HT was reduced, significantly in chronic hypoxic rats where vasoreactivity was increased; HPV and AI responses were maintained (table 3⇑).
In Experiment 1, where AMI was given i.p. (table 2⇑) there was no unequivocal evidence for pulmonary hypertension. Mean Ppa was raised but not significantly. The raised RV/LV+S ratio was most likely due to the reduction in LV weight as RV weight was not increased. Although there was no direct evidence, it is possible that the poor condition of the treated rats, with abdominal distension due to retained faeces, might have led to low cardiac output and Psys. This may have led to the absence of pulmonary hypertension. However, in Experiment 3 (table 3⇑), where AMI was given by gavage and the rats were in better condition, there was also no evidence of right ventricular hypertrophy or muscularisation of pulmonary arterioles in the normoxic group. Additional experiments were performed with concomitant chronic hypoxia, which itself causes pulmonary hypertension and is an accompanying feature of ARDS following TCA overdose. In the chronic hypoxic groups the development of pulmonary hypertension was confirmed by the presence of RV hypertrophy and vascular remodelling, which was unaltered by chronic AMI.
Although the dose regime in Experiments 1 and 3 were similar, the possibility that absorption is more rapid from the peritoneum than the gut, such that the pharmocokinetic profile may have been different leading to a “lower” dosage in the latter, must be considered.
Chronic treatment with iprindole
After iprindole treatment there was clear evidence of pulmonary hypertension with a significantly raised Ppa in Experiment 4a (table 4⇑). In both Experiments 4a and b, where the dosage was identical but work was carried out in Prague (a) and Sheffield (b), there was evidence of right ventricular hypertrophy (RV/LV+S) although this was complicated by lower absolute LV weight associated with reduced body weight in Experiment 4b. However, in both experiments body growth was reduced in the iprindole treated rats with no change in LV/BW. Histological studies were not performed, but Vijeyaratnam and Corrin 19 showed oedema, swelling of the endothelium and epithelium and macrophage infiltration in rats treated chronically with large doses of iprindole.
Anorectic drugs
In a parallel study performed by this group on two anorectic drugs (fenfluramine and chlorphentermine) with similar chemical and biochemical properties to tricyclic antidepressants 3, it was shown that given acutely, they also lead to persistent rises in pulmonary artery pressue. They modified angiotensin converting enzyme activity and pressor effects of serotonin and noradrenaline. There is evidence that both tricyclic antidepressants and fenfluramines inhibit potassium channels 20, 21, a postulated mechanism for fenfluramine-induced pulmonary vasoconstriction and development of pulmonary hypertension 21, 22. Analysis of the similarities and differences between anorectics and tricyclic antidepressants may increase understanding of drug-induced damage to the pulmonary circulation.
- Received March 14, 2001.
- Accepted December 18, 2001.
- © ERS Journals Ltd
References









